
Abstract
Diabetic cardiomyopathy (DbCM) is one of the most common vascular complications of diabetes, and can cause heart failure and threaten the life of patients. The pathogenesis is complex, and key genes have not fully identified. In this study, bioinformatics analysis was used to predict DbCM-related gene targets. Published datasets from the NCBI Gene Expression Omnibus with accession numbers GSE62203 and GSE197850 were selected for analysis. Differentially expressed genes (DEGs) were identified by the online tool GEO2R. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the DAVID online database. Protein–protein interaction network construction and hub gene identification were performed using STRING and Cytoscape. We used 30 mM and 1 μM hydrocortisone-stimulated AC16 cells as an in vitro model of diabetic cardiomyopathy. Quantitative real-time PCR (qRT-PCR) was performed to validate the expression levels of hub genes. A total of 73 common DEGs were identified in both datasets, including 47 upregulated and 26 downregulated genes. GO and KEGG pathway enrichment analyses revealed that the DEGs were significantly enriched in metabolism, hypoxia response, apoptosis, cell proliferation regulation, and cytoplasmic and HIF signalling pathways. The top 10 hub genes were LDHA, PGK1, SLC2A1, ENO1, PFKFB3, EGLN1, MYC, PDK1, EGLN3 and BNIP3. In our in vitro study, we found that PGK1, SLC2A1, PFKFB3, EGLN1, MYC, EGLN3 and BNIP3 were upregulated, ENO1 was downregulated, and LDHA was unchanged. Except for PGK1 and ENO1, these hub genes have been previously reported to be involved in DbCM. In summary, we identified DEGs and hub genes and first reported PGK1 and ENO1 in DbCM, which may serve as potential candidate genes for DbCM targeted therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic cardiomyopathy (DbCM) is a specific type of cardiomyopathy, that is independent of hypertension, coronary artery disease and valvular heart disease. It is a primary lesion of the myocardium caused by chronic hyperglycemia. DbCM is initially characterized by diastolic dysfunction, increased atrial filling, prolonged isovolumic diastolic time, increased superior ventricular premature beat, increased left ventricular end-diastolic pressure, and decreased left ventricular end-diastolic volume, resulting in decreased left ventricular ejection fraction and cardiac output. As the disease progresses, patients experience systolic dysfunction, cardiac remodelling, myocardial fibrosis, myocardial hypertrophy, arrhythmia, and cardiac hypertrophy and finally progress to chronic heart failure [1,2,3]. There are still some difficulties in the clinical diagnosis of diabetic cardiomyopathy. Some researchers have proposed new biomarkers, such as glycosylated hemoglobin, glucose, low density lipoprotein, insulin-like growth factor binding protein 7 and transforming growth factor β1, which can help clinical diagnosis [4]. The pathogenesis of DbCM is very complicated and mainly involves disorders of glucose and lipid metabolism, oxidative stress, apoptosis and insulin resistance [2], but the key genes and molecular mechanisms have not been fully elucidated.
Disease models are essential for understanding cardiovascular pathogenesis and developing new therapeutic approaches. At present, research on DbCM focuses on cell and animal models, and human studies are lacking due to the particularity of heart samples. In recent years, human-induced pluripotent stem cell-derived cardiomyocytes (hiPSCs-CMs) have provided an excellent experimental model for cardiovascular diseases, such as arrhythmogenic cardiomyopathy [5] and hypertrophic cardiomyopathy [6]. Since these cells express cardiomyocyte-specific markers, they present typical electrophysiological characteristics of cardiomyocytes and respond to catecholamines and cholinergic drugs. Thus, hiPSCs-CMs have often been used to build cell models of cardiomyopathy. Some studies have reported that hiPSC-CMs develop insulin resistance under specific culture conditions, indicating that hiPSC-CMs have great potential in DbCM research [7].
In this study, differentially expressed genes (DEGs) between DbCM and control samples were first identified from NCBI Gene Expression Omnibus (GEO) datasets (GSE62203 and GSE197850), and the overlapping DEGs were reserved for downstream analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to explore the biological processes and molecular pathways of DEGs. Protein–protein interaction (PPI) networks were constructed to identify the hub genes among the DEGs. Overall, this study will provide new molecular targets and theoretical evidence for clinical treatment of DbCM.
Materials and methods
General information
hiPSCs-CMs datasets related to DbCM were collected from the NCBI GEO repository with accession numbers GSE62203 and GSE197850.
Differentially expressed genes (DEGs) identification
DEGs between the disease model and control groups from the GSE197850 dataset were directly downloaded, while the DEGs in GSE62203 were screened by using GEO2R. Genes with P < 0.05 and absolute fold change (FC) > 2 were considered significantly differentially expressed. The DEGs that were consistently up or downregulated in both datasets were used for further analysis.
Functional and pathway enrichment analysis
To investigate the role of the DEGs, GO and KEGG signalling pathway enrichment analyses were performed by using the DAVID online database. The GO analysis consisted of biological processes, cellular components and molecular functions. Terms with P < 0.05 were considered significantly enriched.
Protein–protein interaction (PPI) network construction
PPI networks of the common DEGs were constructed through STRING. The minimum needed interaction score was 0.4 and the retained PPI networks were visualized by Cytoscape.
Hub gene identification
The PPI networks from the STRING database were imported into Cytoscape, and the MNC algorithm was used to identify hub genes.
Evaluation of hub genes
ROC curves of hub genes were generated using GraphPad Prism 8.0. To estimate the specificity and sensitivity of hub genes for the diagnosis of DbCM, we performed receiver operating characteristic (ROC) analysis of each hub gene. The area under the curve (AUC) value was calculated to quantify the diagnostic performance. Genes with AUC values greater than 0.7 were considered as potential diagnostic indicators of DbCM. A two-sample t-test was used to compare the gene expression levels of DbCM and normal samples.
Cell culture
The human myocardial cell line AC16 was used in this study and was maintained in DMEM (Gibco), supplemented with 20% foetal bovine serum (Pricella), 100 U/ml penicillin (Solarbio) and 0.1 mg/ml streptomycin (Solarbio) at 37 ℃ in an atmosphere of 5% CO2. To establish a diabetic cardiomyopathy model, cells were treated with different conditions of (a) 25 mM glucose and 1% DMSO; (b) 30 mM glucose and 1 μM hydrocortisone (MedChemExpress) and cultured for 24 h.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was purified using TRIzol reagent according to the manufacturer’s instructions (Invitrogen) and processed for cDNA synthesis using the HiScript Q RT SuperMix kit (Vazyme). cDNA was analysed by qRT-PCR with ChamQ Universal SYBR qPCR Master Mix (Vazyme) using the LightCycler 480 qPCR analysis system (Roche). The mRNA expression levels of target genes were normalized to GAPDH. The primers used in our study are listed in the Supplementary Table.
Statistical analysis
Using GAPDH as the housekeeping gene, we calculated the relative changes in the expression level of target genes in DbCM AC16 cells compared with the control group by 2-△△Ct, normalized the control group, repeated the experiment for six times, and detected the difference of components by unpaired t-test. P < 0.05 was considered to be statistically significant.
Results
The co-DEGs of the GSE62203 and GSE197850 datasets
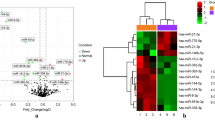
A total of 364 DEGs, including 188 upregulated and 176 downregulated genes, were identified in the GSE62203 dataset. A total of 1400 DEGs, including 569 upregulated and 831 downregulated genes (Table 1), were directly obtained from GSE197850. A total of 78 DEGs were identified in these two gene expression profiles (Fig. 1a). Of these 73 genes were differentially expressed simultaneously (called co-DEGs), including 47 upregulated genes (64.38%) and 26 downregulated genes (35.62%), and were retained for downstream analysis (Fig. 1b). The expression levels of co-DEGs in GSE197850 and GSE62203 are shown in Fig. 1c and Fig. 1d, respectively.

DEGs in GSE62203 and GSE197850. (a) Venn diagram of the overlapping DEGs in GSE62203 and GSE197850, (b) Pie chart show the proportions of upregulated and downregulated genes shared in GSE62203 and GSE197850, (c, d) Heat map of the expression levels of 73 co-DEGs in the GSE197850 (c) and GSE62203 (d)
GO and KEGG pathway enrichment analysis of co-DEGs
To explore the function of co-DEGs, GO and KEGG signalling pathway enrichment analyses were performed on 73 co-DEGs using the DAVID online database. Ninety-six terms were obtained from GO analysis. In the cellular components category, the DEGs were mainly enriched in cell surface, exosomes, mitochondria, endoplasmic reticulum and cytoplasm (Fig. 2a). In the biological processes category, the DEGs were involved in hypoxia response, glycolipid metabolism, cell proliferation and growth, apoptosis and myocardial contraction regulation (Fig. 2b); In the molecular functions category, the DEGs were mainly enriched in iron ion binding and hormone activity (Fig. 2c). KEGG analysis showed that the DEGs were mainly enriched in the HIF signalling pathway, amino acid anabolism and carbon metabolism (Fig. 2d).

GO and KEGG signalling pathway enrichment results. (a) Cellular components analysis of co-DEGs, (b) Biological processes analysis of co-DEGs, (c) Molecular functions analysis of co-DEGs and (d) KEGG signalling pathway analysis of co-DEGs
PPI network construction
The online tool STRING was used to construct PPI networks of co-DEGs. In the network, there were 69 nodes and 176 edges. The average node degree was 5.1 and the local clustering coefficient was 0.507, which indicates that the PPI enrichment was statistically significant (Fig. 3).

PPI networks of co-DEGs
Hub gene identification
The hub genes in the PPI networks were identified by CytoHubba (Fig. 4a). The top 10 hub genes including LDHA, PGK1, SLC2A1, ENO1, PFKFB3, EGLN1, MYC, PDK1, EGLN3 and BNIP3 were determined by the MNC algorithm (Fig. 4b and Table 2).

Protein interaction map of the top 10 hub genes. (a) A PPI network of co-DEGs constructed using Cytoscape and STRING, (b) The top 10 hub genes were identified by CytoHubba using MNC algorithm
Evaluation of hub genes
In the GSE62203 and GSE197850 datasets, except for the MYC gene, the mRNA expression levels of the hub genes were higher in the DbCM group compared to the control group.. To further explore the diagnostic value of these 10 genes in DbCM, the area under the curve (AUC) of receiver operating characteristic (ROC) was calculated to judge the diagnostic value of the 10 hub genes. The AUC of each hub gene was greater than 0.7, indicating that 10 hub genes might play a role in clinical diagnosis of DbCM (Fig. 5a and 6a).

Validation of hub genes in the GSE62203 dataset. ROC analysis of hub genes in DbCM

Validation of hub genes in the GSE197850 dataset. ROC analysis of hub genes in DbCM
Validation of the expression levels of hub genes in DbCM by qRT-PCR
To further verify the expression levels of the top 10 hub genes in DbCM, we established an in vitro model of DbCM by using the AC16 cell line and performed qRT-PCR to determine their mRNA expression levels. The results showed that the mRNA expression levels of the PFKFB3, MYC, EGLN3, EGLN1, BNIP3, and PGK1 genes were significantly increased in the DbCM group compared with the control group, which was consistent with the above results. In contrast, the mRNA expression levels of ENO1 was significantly decreased in the DbCM group. However, the mRNA expression levels of LDHA and SLC2A1 were slightly increased in the DbCM group compared with the control group, but this difference was no significant (Fig. 7a, b).

mRNA expression levels of the hub genes were quantified by qRT-PCR. (a) Agarose gel electrophoresis image of qRT-PCR product. Each band represented an individual sample. (b) Bar charts depicting the expression of hub genes in DbCM and Control group
Discussion
Diabetic cardiomyopathy is one of the most common vascular complications in patients with diabetes. According to a survey of the 2021 International Diabetes Federation, cardiovascular complications of diabetes including DbCM are the most critical factors causing morbidity and mortality in patients with diabetes. The pathogenesis of DbCM mainly involves disorders of glucose and lipid metabolism, oxidative stress, endothelial cell dysfunction, inflammation and apoptosis. Thus, it is complex and has not been fully elucidated. Therefore, it is critical to find the key therapeutic target for DbCM. In this study, we analysed the DEGs in two DbCM datasets using hiPSCs-CMs to construct DbCM models, and performed GO and KEGG signalling pathway enrichment analyses. A total of 10 hub genes that may play important roles in DbCM were screened.
Establishing a cell model that can simulate the pathophysiological state of the human body is an important method in medical research. Although neonatal rat cardiomyocytes, the rat cardiomyocyte cell line H9c2, and the mouse cardiomyocyte cell line HL-1 are frequently employed in cardiovascular disease research [8], rat, mouse and human hearts differ greatly in terms of gene expression and energy metabolism during disease states [9]. Therefore, it is more reliable to construct a DbCM model using human-derived cardiomyocytes. Human induced pluripotent stem cells (hiPSCs) exhibit the properties of self-renewal and pluripotency. Moreover, cardiomyocytes differentiated from hiPSCs have great advantages in the field of cardiovascular diseases and have been widely used in the past decade. Therefore, the datasets of the diabetic cardiomyopathy model constructed by hiPSCs-CMs were selected for this study.
The expression levels of 10 DEGs in the two datasets were consistent, and the AUC was greater than 0.7, indicating high diagnostic value. LDHA is a subtype of lactate dehydrogenase, which is one of the key enzymes in glycolysis, and can catalyse pyruvate to lactate. Studies have shown that increased LDHA activity can promote lactate levels and lactate-pyruvate conversion in patients with diabetes [10]. However, the expression levels of LDHA were reduced in the hearts of STZ-induced diabetic mice, possibly due to the inhibition of glycolysis [11]. Interestingly, we verified changes in the mRNA levels of LDHA by qRT-PCR and found no statistically significant difference between the DbCM and control groups. This may be due to the different maturation of the cell lines. SLC2A1, also known as GLUT1, is involved in the regulation of glycometabolism, and its expression levels were increased in hiPSCs-CMs stimulated by high glucose, which is consistent with the results of our study. A previous study revealed that empagliflozin induced cardiac dysfunction by upregulating the expression of SLC2A1 [12]. Fructose-2-kinase 6-phosphate/fructose-2, 6-diphosphatase 3 (PFKFB3), regulates the rate of glycolysis by controlling the levels of fructose-2, 6-diphosphate (F-2,6-BP), which is believed to be an effective allosteric activator of fructose-1-kinase 6-phosphate (PFK1). PFK1 is a key rate-limiting enzyme in glycolysis that catalyses the conversion of F-6-P to F-1, 6-BP and is considered to be a major rate-limiting step in glycolytic flux [13]. In astrocytes, PFKFB3 is maintained a low level through proteasome-mediated ubiquitination degradation, ensuring the predominance of the pentose phosphate pathway in glucose metabolism. Therefore, the level of NADPH is increased, which contributes to oxidative damage repair [14, 15]. The mRNA expression level of PFKFB3 is upregulated, the rate of glycolysis is increased, and the inflammatory response is enhanced in endothelial cells in a high glucose environment [16, 17]. Consistent with previous studies, we found that the mRNA expression level of PFKFB3 was elevated in the DbCM group, which was consistent with previous studies. Therefore, we speculate that PFKFB3 may play a role in the pathogenesis of DbCM. PGK1 and ENO1 are also key enzymes for glycolysis. PGK1 is an oncogene in cancer occurrence and progression. It can promote cell growth and proliferation [18]. Additionally, PGK1 is a key metabolic enzyme in the glycolysis process. It functions to catalyze the conversion of 1, 3-diphosphoglyceric acid (1,3-BPG) to 3-phosphoglyceric acid (3-PG) and produces the first ATP in the glycolysis pathway [19]. Our study first identified elevated PGK1 expression levels in DbCM. This may provide a new molecular target for DbCM research. Similarly, ENO1 is also involved in the processes of division, proliferation, metastasis and apoptosis of tumour cells [20]. The results of bioinformatics analysis showed that the expression level of ENO1 in DbCM was increased, but in vitro experiments showed that the expression level of ENO1 in the DbCM model was decreased. The role of ENO1 in DbCM may need further clarification in the future. Therefore, LDHA, SLC2A1, PFKFB3, PGK1 and ENO1 may be potential therapeutic targets for DbCM, especially PGK1 and ENO1.
Prolyl hydroxylase (EGLN) oxygen-dependent hydroxylation of specific proline residues in hypoxia-inducible factor subunits participates in the oxygen-sensing pathway. EGLN1 and EGLN3 are two important members of the EGLN protein family that have been reported to play an important role in the pathogenesis and progression of DbCM. EGLN1, also known as PHD2, was significantly increased in the heart tissue of db/db mice. Overexpression of angiotensin-1 may rescue the impairment of angiogenesis in diabetes by inhibiting PHD2 expression [21]. The expression levels of EGLN3, also known as PHD3, were significantly increased in DbCM rats. Knockdown of EGLN3 can improve cardiac function and inhibit fibrosis and apoptosis in DbCM rats. The expression levels of EGLN3 were significantly increased when H9c2 cells were exposed to high glucose conditions. The decrease in EGLN3 levels could inhibit the increase in apoptosis caused by high glucose conditions [22]. Similarly, the results of GO and KEGG analyses of DEGs in our study showed that EGLN1 and EGLN3 play important roles in DbCM by participating in the cellular hypoxia response and HIF-1 signalling pathway. Therefore, our study confirmed the increased expression of EGLN1 and EGLN3 in DbCM, supporting the conclusions of previous studies.
Apoptosis is one of the key molecular mechanisms in the pathogenesis and progression of DbCM. Bcl-2 interacting protein 3 (BNIP3) is a pro-apoptotic protein, and the levels of BNIP3 were significantly increased in DbCM rat hearts. This funding is consistent with the conclusions of our study. The increased expression levels of BNIP3 in H9c2 cells induced by high glucose stimulation can lead to increased apoptosis. This indicates that BNIP3, one of the important molecules of apoptosis, is involved in the progression of DbCM [23]. MYC is a proto-oncogene that expresses a nuclear phosphorylated protein and plays an important role in the cell cycle and apoptosis. Studies have shown that the expression level of c-MYC in the heart of T2D rats was increased [24]. Interestingly, after high glucose stimulation in H9c2 cardiomyocytes, c-myc protein expression levels were significantly decreased and the cell proliferation cycle was arrested [25]. In our study, the mRNA expression levels of MYC were also significantly increased, possibly due to its involvement in apoptosis via the Wnt/ beta-catenin pathway [26]. These seemingly contradictory results may be mainly due to different mouse strains and different disease stages simulated by the two disease models. The C-myc protein is involved in various cellular pathophysiological processes and can promote the pathogenesis of DbCM. Thus, it may be an important molecular target for further study in the future. PDK1, a serine/threonine protein kinase, inhibits apoptosis by increasing the phosphorylation of AKT. Hou and Lin et al. discovered that the phosphorylation of PDK was decreased in DM rats and mice [15, 27]. However, the low expression level of PDK1 in AC16 makes it difficult to detect PDK1 mRNA by qRT-PCR methods.
There are some limitations of this study. First, we used AC16 cells to verify the expression of hub genes in our study. Although AC16 is a human cardiac cell line and is widely used in human cardiology research, AC16 still differs from hiPSCs-CMs in terms of maturity and gene expression. Second, only 16 samples from both datasets were used in our study for analysis. In the future, we will expand the sample size for further analysis. Third, PDK1 expression was low in AC16 cells, and thus, its role in DbCM cannot be verified. In the future, PDK1 overexpressing AC16 cells could be used to confirm the expression changes of PDK1 in the diabetic state.
Conclusions
In this study, 73 genes with altered expression in DbCM were screened and were mainly found to be enriched in glucose and lipid metabolism disorders, hypoxia response, cell apoptosis, cell proliferation regulation, and cytoplasmic and HIF signalling pathways. Furthermore, we identified 10 hub genes including LDHA, PGK1, SLC2A1, ENO1, PFKFB3, EGLN1, MYC, PDK1, EGLN3 and BNIP3 from the DEGs. In vitro studies, we found that the mRNA expression levels of PGK1, PFKFB3, MYC, EGLN1, EGLN3 and BNIP3 were upregulated, LDHA and SLC2A1 were unchanged, and ENO1 was downregulated in the DbCM group. Our study first found PGK1 and ENO1 were involved in DbCM, and the effect of PDK1 and MYC in DbCM needs to be verified and clarified. Our study provided new molecular targets and theoretical basis for the clinical diagnosis of DbCM, and offered a new perspective for future DbCM research.
Data Availability
The open-access datasets are available through the following URL: GSE62203 (https://www.ncbi.nlm.nih.gov/gds/?term=GSE62203) and GSE197850. (https://www.ncbi.nlm.nih.gov/gds/?term=GSE197850). All data generated or analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- DbCM:
-
Diabetic cardiomyopathy
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- hi-PSCs:
-
Human induced pluripotent stem cells
- hiPSCs-CMs:
-
Human induced pluripotent stem cell-derived cardiomyocytes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–protein interaction
References
Ibrahim E-SH, Dennison J, Frank L et al (2021) Diastolic cardiac function by MRI-imaging capabilities and clinical applications. Tomography 7:75
Avagimyan A, Popov S, Shalnova S (2022) The pathophysiological basis of diabetic cardiomyopathy development. Curr Probl Cardiol 47(9):101156
Tadic M, Cuspidi C, Calicchio F et al (2020) Diabetic cardiomyopathy: how can cardiac magnetic resonance help? Acta Diabetol 57:1027–1034
Kumric M, TicinovicKurir T, Borovac JA (2021) Role of novel biomarkers in diabetic cardiomyopathy. World J Diabetes 12:685–705
Chua CJ, Morrissette-McAlmon J, Tung L et al (2023) Understanding arrhythmogenic cardiomyopathy: advances through the use of human pluripotent stem cell models. Genes (Basel) 14(10):1864
Fujiwara Y, Miki K, Deguchi K et al (2023) ERRγ agonist under mechanical stretching manifests hypertrophic cardiomyopathy phenotypes of engineered cardiac tissue through maturation. Stem Cell Reports S2213–6711(23):00362–00364
Purnama U, Castro-Guarda M, Sahoo OS, Carr CA (2022) Modelling diabetic cardiomyopathy: using human stem cell-derived cardiomyocytes to complement animal models. Metabolites 12(9):832
Onódi Z, Visnovitz T, Kiss B, Hambalkó S, Koncz A, Ágg B et al (2022) Systematic transcriptomic and phenotypic characterization of human and murine cardiac myocyte cell lines and primary cardiomyocytes reveals serious limitations and low resemblances to adult cardiac phenotype. J Mol Cell Cardiol 165:19–30
Linscheid N, Santos A, Poulsen PC, Mills RW, Calloe K, Leurs U et al (2021) Quantitative proteome comparison of human hearts with those of model organisms. PLoS Biol 19(4):e3001144
Li H, Li X, Guo J, Wu G, Dong C, Pang Y et al (2020) Identification of biomarkers and mechanisms of diabetic cardiomyopathy using microarray data. Cardiol J 27(6):807–816
Wang JY, Ma D, Luo M, Tan YP, Ou Zhong, Tian G, et al. Effect of spermidine on ameliorating spermatogenic disorders in diabetic mice via regulating glycolysis pathway. Reprod Biol Endocrinol. 2022;20(1):45.
Ng KM, Lau YM, Dhandhania V, Cai ZJ, Lee YK, Lai WH et al (2018) Empagliflozin ammeliorates high glucose induced-cardiac dysfunction in human iPSC-derived cardiomyocytes. Sci Rep 8(1):14872
Pilkis S, Claus T, Kurland I, Lange A (1995) 6-Phosphofructo-2-kinase/ fructose-2,6-bisphosphatase: a metabolic signaling enzyme. Annu Rev Biochem 64:799–835
Cavalier MC, Kim SG, Neau D, Lee YH (2012) Molecular basis of the fructose-2,6-bisphosphatase reaction of PFKFB3: transition state and the C-terminal function. Proteins 80:1143–1153
Hou N, Mai Y, Qiu X, Yuan W, Li Y, Luo C et al (2019) Carvacrol attenuates diabetic cardiomyopathy by modulating the PI3K/AKT/GLUT4 pathway in diabetic mice. Front Pharmacol 12(10):998
Atawia RT, Batori RK, Jordan CR et al (2023) Type 1 diabetes impairs endothelium-dependent relaxation via increasing endothelial cell glycolysis through advanced glycation end products, PFKFB3, and Nox1-mediated mechanisms. Hypertension 80(10):2059–2071
Song C, Wang S, Fu Z et al (2022) IGFBP5 promotes diabetic kidney disease progression by enhancing PFKFB3-mediated endothelial glycolysis. Cell Death Dis 13(4):340
He Y, Wang X, Lu W, Zhang D, Huang L, Luo Y et al (2022) PGK1 contributes to tumorigenesis and sorafenib resistance of renal clear cell carcinoma via activating CXCR4/ERK signaling pathway and accelerating glycolysis. Cell Death Dis 13(2):118
Li X et al (2016) Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell 61:705–719
Zuo J, Wang B, Long M, Gao Z, Zhang Z, Wang H et al (2018) The type 1 transmembrane glycoprotein B7–H3 interacts with the glycolytic enzyme ENO1 to promote malignancy and glycolysis in HeLa cells. FEBS Lett 592(14):2476–2488
Chen JX, Stinnett A (2008) Ang-1 gene therapy inhibits hypoxia-inducible factor-1alpha (HIF-1alpha)-prolyl-4-hydroxylase-2, stabilizes HIF-1alpha expression, and normalizes immature vasculature in db/db mice. Diabetes 57(12):3335–3343
Xia Y, Gong L, Liu H, Luo B, Li B, Li R et al (2015) Inhibition of prolyl hydroxylase 3 ameliorates cardiac dysfunction in diabetic cardiomyopathy. Mol Cell Endocrinol 5(403):21–29
Zhou W, Yang J, Zhang DI, Li F, Li G, Gu Y, Luo M (2015) Role of Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 in myocardial cells in diabetes. Exp Ther Med 10(1):67–73
Rami M, Rahdar S, Ahmadi Hekmatikar A, AwangDaud DM (2023) Highlighting the novel effects of high-intensity interval training on some histopathological and molecular indices in the heart of type 2 diabetic rats. Front Endocrinol (Lausanne) 19(14):1175585
Zhao C, Shen Q (2018) Overexpression of small ubiquitin-like modifier 2 ameliorates high glucose-induced reductions in cardiomyocyte proliferation via the transforming growth factor-β/Smad pathway. Mol Med Rep 18(6):4877–4885
Zhou W, Yang J, Zhang DI, Li F, Li G, Gu Y et al (2015) Role of Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 in myocardial cells in diabetes. Exp Ther Med 10(1):67–73
Lin G, Brownsey RW, Macleod KM (2014) Complex regulation of PKCβ2 and PDK-1/AKT by ROCK2 in diabetic heart. PLoS ONE 9(1):e86520
Acknowledgements
None.
Funding
This work was supported by National College Students' Innovative Entrepreneurial Training Plan Program (202210062018), CAMS Innovation Fund for Medical Sciences (CIFMS) (approval No. 2021-I2M-1–073, 2022-I2M-C&T-B-091), Tianjin Natural Science Foundation (approval No. 20JCZDJC00410, TJWJ2022MS001), Sansheng TCP Young Research Funding (approval No. 57) and National Natural Science Foundation of China (approval No. 82170217, 82070192, 81670171).
Author information
Authors and Affiliations
Contributions
Shuo An, Hongchen Bi and Xiaoli Luo wrote the main manuscript text. Caiying Zhu and Min Wang prepared Figs.1-7. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethical approval and consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
An, S., Bi, H., Luo, X. et al. Identification of key genes of diabetic cardiomyopathy in hiPSCs-CMs based on bioinformatics analysis. Mol Cell Biochem (2024). https://doi.org/10.1007/s11010-023-04915-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11010-023-04915-9