
Abstract
The progression of atherosclerotic plaque is accelerated by death of foam cells during the development of the plaque. There are several forms of foam cell death, such as autophagy and ferroptosis forms of cell death together are commonly predominant. Therefore, it is particularly important to study the crosstalk between various forms of cell death in atheroscler and ferroptosis. Although there is a dominant form of cell death that plays a role in the disease, motic plaques. Nuclear factor NF-E2-related factor (Nrf2) has been considered as a major regulator of antioxidant in previous studies, but recent studies have revealed that insufficient cellular autophagy can turn off Nrf2-mediated antioxidant defense while initiating Nrf2-manipulated iron deposition and lipid peroxidation, leading to the development of iron ferroptosis. The present experiment aimed to explain the regulatory mechanism between autophagy and ferroptosis through Nrf2. In this experiment, differentiated human THP-1 macrophages were used, which were treated with ox-LDL into foam cells with the addition of the autophagy inhibitor chloroquine (CQ), the inhibitor of Nrf2 (ML385), the promoter of Nrf2 (t-BHQ), and the inhibitor of ferroptosis (Liproxstatin-1), and the expression levels of autophagy-related proteins p62 and LC3, as well as Nrf2 and ferroptosis-related proteins xCT and GPX4 by WB, foam cell survival by CCK8, and intracellular reactive oxygen levels by Flow cytometry analysis and fluorescence microscopy. The effect of autophagy through Nrf2 on ferroptosis in foam cells was determined. The results revealed that insufficient autophagy in CQ-induced foam cells could lead to foam cell death in atherosclerotic plaques, and the cause of cell death was that insufficient autophagy in foam cells turned off the positive effect of Nfr2 antioxidant, initiated the negative effect of Nrf2 to promote intracellular reactive oxygen species production, and this negative effect promoted ferroptosis in foam cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis (AS) is a progressive lesion with age, and cardiovascular diseases caused by AS remain the leading cause of death in humans worldwide, seriously threatening people's health and lives [1]. To date, a large number of domestic and international articles have investigated the role of cell death pathways in AS, especially foam cells formed by macrophages play an important role in promoting the progression of AS [2]. Among the various forms of cell death, ferroptosis and autophagy are the hot spots of research in recent years. Ferroptosis was initially considered to be an iron-dependent death distinct from other programmed deaths, and some studies have shown that ferroptosis plays an important role in AS [3].
Autophagy is a subcellular process that facilitates the turnover of intracellular material, and the initial understanding of autophagy in AS was limited to its favorable aspect of impeding the disease process [4]. A large number of recent studies have confirmed that autophagy has a bidirectional role in the regulation of AS during the AS process. Autophagy at the basal level protects the cells of AP from various stressful environments [5]. In contrast, dysregulation of autophagy as well as excessive levels of autophagy during late AS can promote the formation and rupture of AP [6]. In late AS, severe stress can cause defects in the formation of autophagosomes or in the fusion and degradation of autophagosomes with lysosomes. Defective autophagy can disrupt the balance of lipid metabolism, promote lipid deposition in the vessel wall, and significantly increase AP formation [7]. However, excessive autophagy can lead to the release of inflammatory factors and severe oxidative stress as well as the formation of autophagic wax-like bodies, which are complexes of oxidized lipids and proteins that are not digested by lysosomes and also limit the binding of autophagosomes to lysosomes, reduce collagen fiber synthesis in smooth muscle cells leading to thinning of the fibrous cap, promote thrombosis by endothelial cell death and death of macrophages Increased AP load and instability [8]. It has been shown that knockdown of autophagy-related gene5 (ATG5) and autophagy-related gene7 (ATG7) reduces intracellular iron levels and lipid peroxidation thereby inhibiting ferroptosis induced by the ferroptosis activator Erastin [9]. Therefore, ferroptosis is considered to be an autophagy-dependent death under specific circumstances, but the role of autophagy and ferroptosis in AS has not been explored.
Nrf2 has been regarded as a major regulator of antioxidant in previous studies, and Nrf2 inhibits ferroptosis by promoting iron storage and reducing iron uptake and reactive oxygen species(ROS) production [10] Zang et al. [11] found that diabetes causes insufficient autophagy in cardiomyocytes, which shuts down Nrf2-mediated antioxidant defense, while initiating Nrf2-mediated iron deposition and lipid peroxidation, leading to the development of ferroptosis. And the ferroptosis inducer Eaststin inhibited the autophagic flux, suggesting that autophagy inhibition may play an important role in Nrf2-mediated ferroptosis. Nrf2 is commonly expressed in the cardiovascular system, and Nrf2 is a key regulator in maintaining cardiovascular homeostasis by inhibiting oxidative stress [12]. However, Mimura et al. [13] found that Nrf2 showed both pro-atherogenic AS effects and anti-AS effects in experimental animal models. It was found that defective autophagy can trigger non-classical activation of Nrf2 leading to sustained activation of Nrf2, and that abnormal activation of Nrf2 exacerbates cellular inflammatory responses, promotes cell death, and is pathogenic [14]. It has also been shown in numerous studies that upregulation of Nrf2 can lead to the development of autophagy deficiency [15, 16]. Therefore, we hypothesized that in the case of insufficient autophagy in advanced AS, Nrf2 likely also undergoes non-classical activation, and a vicious cycle between non-classical activation of Nrf2 and insufficient autophagy promotes foam cell death. Nrf2 may be expected to link autophagy and ferroptosis in the disease to become a new target for AS therapy.
Materials and methods
Cell culture and differentiation
The cells used in this experiment were human-derived THP-1 cell line, provided by BeNa Culture Collection (BNCC, Beijing, China).The cells were cultured in RPMI1640 (Hyclone, USA): fetal bovine serum (FBS) (Hyclone, USA) in a 9:1 ratio of complete medium, and the content of the culture medium in the dishes was adjusted to approximately 7–9 ml according to the number of cells, and the cells were cultured in a 5% CO2, 37 °C cell culture incubator. According to the growth status of the cells, the medium was changed every 2–3 days. After the cells reached the logarithmic growth phase, the cells were diluted with 2% FBS RPMI1640 to 1 × 106/ml and spread in 6-well plates or 96-well plates. The cells were treated with 100 ng/ml phorbol-12-myristate-13-ace-tate (PMA) (Sigma, USA) for 48 h to induce THP-1 monocytes into macrophages.
Construction of foam cell model
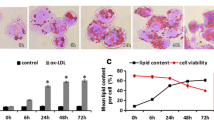
Cells grown in logarithmic phase (cell density reached 5 × 105/ml) were taken, diluted to 1 × 106/ml with 2% FBS RPMI1640 and spread in 6-well plates or 96-well plates, treated with PMA for 24 h. After induction into macrophages, the old medium was discarded, and new medium with 2% FBS RPMI1640 was added with 25, 50, 75 µg/ml concentrations ox-LDL to treat the THP-1 macrophages for 48 h, then observed by microscopy, and the ox-LDL concentration of 50 µg/ml, which had less cell damage and higher success rate of foam cell construction, was selected for subsequent experiments.
Western blot (WB)
The treated cells in the 6-well plate were washed three times with PBS after discarding the medium. An appropriate amount of prepared protein lysate (RIPA:sodium orthovanadate:PMSF in the ratio of 200:1:1) was added to each well, and the total protein was extracted and crushed with an ultrasonic crusher for 1 min. The protein concentration was measured with a BCA kit (Solarbio, Beijing, China). An equal amount of SDS-PAGE electrophoresis was performed with 30 mg of egg added to each well. The run was transferred to cellulose acetate membrane and incubated for 16–20 h at 4 °C. The primary antibody was diluted as follows: LC3B (abcam, USA, dilution ratio 1:3000), β-actin (abcam, USA, dilution ratio 1:5000), p62 (abcam, USA, dilution ratio 1:3000), xCT (abcam, USA, dilution 1:3000), GPX4 (abcam, USA, dilution 1:2000), and Nrf2 (abcam, USA, dilution 1:2000). Afterwards, the secondary antibodies were incubated for 1 h on a room temperature shaker at the following dilutions: goat anti-mouse IgG (abcam, USA, dilution ratio 1:10,000) and goat anti-rabbit IgG (abcam, USA, dilution ratio 1:50,000).The incubated strips were exposed with an imager (Tanon 5200) and the exposed strips were measured in grayscale using Image J.
CCK8 cell activity assay
Cell viability was measured using the Cell Counting Kit-8.Cells treated with different conditions were discarded from the original medium in a 96-well plate, and 100 μl of Cell Counting Kit-8 reagent at a concentration of 10 mg/ml was added to each well, and the cells were incubated in a cell incubator for 5 h after adding the reagent. The optical density (OD) values at 450 nm were measured with an enzyme marker, and each group of experiments was repeated three times, and each result was repeated three times to take the average value as the result.
Intracellular ROS assay
Intracellular ROS levels were measured using the Reactive Oxygen Species Assay Kit. The drug-treated foam cells were digested down with trypsin, and a positive control with the addition of reactive oxygen species and a negative control without stimulation were set up. Dilute the fluorescent probe DCFH-DA with serum-free culture medium according to 1:1000, remove the cell culture medium and add 1 ml of diluted DCFH-DA so that it can adequately cover the cells. incubate the cells in a cell culture incubator at 37 °C for 20 min, mix upside down every 3–5 min, and wash the cells three times with serum-free cell culture medium to adequately remove the DCFH-DA that has not entered the cells. Cells loaded with the probe were observed for intracellular ROS levels by fluorescence microscopy, and intracellular ROS levels were quantified by FCM.
Statistical analysis
The experimental data were statistically analyzed using GraphPad Prism 7.0 software. The measurement data were expressed as mean ± standard deviation, firstly, normality test, independent sample t test was used for samples between two groups, and one-way ANOVA was used for samples between multiple groups. P indicates significant difference, and P < 0.05 (two-sided) was considered a statistically significant difference.
Results
Nrf2 was increased by ox-LDL stimulation and xCT and GPX4 were decreased by ox-LDL stimulation
Foam cell formation was a distinctive feature of AS [17]. THP-1 macrophages were treated with 25, 50 and 75 µg/ml of ox-LDL for 48 h, respectively, to induce foam cell formation. The expression of Nrf2, xCT, and GPX4 proteins within the fine was detected by WB. It was found that the expression of Nrf2 increased and the expression of xCT and GPX4 decreased with the increase of ox-LDL concentration (Fig. 1a). It was also found that the expression of Nrf2 in cells treated with 50 µg/ml ox-LDL was significantly higher than that in cells treated with 25 µg/ml ox-LDL (Fig. 1b), and the expression of xCT and GPX4 in cells treated with 25 µg/ml and 50 µg/ml ox-LDL was significantly lower than that in cells not treated with ox-LDL (Fig. 1c, d), demonstrating that Nrf2, xCT, GPX4 protein expression may be correlated with AS.

ox-LDL induces upregulation of Nrf2 and downregulation of xCT and GPX4. a THP-1 macrophages were treated with 25, 50, and 75 µg/ml of ox-LDL for 48 h, respectively, and intracellular Nrf2, xCT, and GPX4 protein expression was detected by WB. b–d Protein expression levels of Nrf2, xCT, and GPX4, respectively. Each band was analyzed and calculated using Image J, and each set of data were obtained from three independent tests and expressed as the mean value. ***P < 0.001, ****P < 0.0001, ns indicates not significant between specific designated groups
Inadequate autophagy of foam cells accelerates foam cell death
In order to simulate the autophagy deficiency of foam cells within the late AS plaques, we added the autophagy inhibitor chloroquine (CQ) to the constructed foam cell model and detected the expression of LC3 and p62, proteins that can determine the autophagy status within the fine by WB. It was found that the level of LC3II/LC3I was significantly reduced in the foam cells with CQ added (Fig. 2a, b), and the expression level of p62 was significantly increased (Fig. 2a, c), which indicated that the foam cell autophagy deficiency was successfully constructed. Cell survival was detected with CCK8 and it was found that the deficiency of autophagy within foam cells could accelerate foam cell death (Fig. 2d). However, the death of foam cells within the AS plaques could increase the instability of the plaques and accelerate the progression of AS [18].

Insufficient autophagy of CQ-induced foam cells leads to reduced survival of foam cells. a THP-1 macrophages treated with 50 µg/ml ox-LDL in 6-well plates for 48 h, followed by 30 µMCQ for 12 h. Intracellular expression of LC3 and p62 was detected by WB. b–c The expression levels of LC3 and p62 were calculated using Image J for each band. Bands were analyzed and calculated, with each data set from three independent tests and expressed as mean values. d THP-1 cells were added to a 96-well plate and induced into macrophages with PMA, and then the cells were divided into 4 groups, and each experiment was repeated three times. The first group was a blank control group. The second group was treated with 30 µM CQ for 12 h after 48 h of culture, the third group was treated with 50 µg/ml ox-LDL for 48 h after 12 h of culture, and the fourth group was treated with 50 µg/ml ox-LDL for 48 h and 30 µM CQ for 12 h. 2% FBS RPMI1640 culture medium was changed every 12 h during this period. After treatment, CCK8 reagent was added and cell activity was detected by enzyme marker. *P < 0.05, ****P < 0.0001, ns indicates not significant between specific designated groups
Insufficient autophagy in foam cells leads to the shutdown of Nrf2 antioxidant action
To investigate the effect of foam cell autophagy deficiency on the role of intracellular Nrf2, we treated foam cells with autophagy inhibitor CQ, Nrf2 promoter t-BHQ and Nrf2 inhibitor ML385. It was found that CQ-induced insufficient autophagy in foam cells could increase the expression of intracellular Nrf2 (Fig. 3a, b), while treatment of foam cells with Nrf2 promoter t-BHQ increased the expression of autophagy-negative protein p62, and treatment of foam cells with Nrf2 promoter t-BHQ decreased the expression of p62 (Fig. 3a, c). The above results suggest a positive feedback regulation of intracellular autophagy deficiency and Nrf2. Insufficient autophagy in foam cells reduced cell survival, and further promotion of Nrf2 expression in the presence of insufficient autophagy in foam cells further reduced cell survival, while inhibition of Nrf2 expression in the presence of insufficient autophagy in foam cells increased cell survival and reversed the reduction in cell survival caused by insufficient autophagy (Fig. 3d). This result indicates that the decrease in cell survival caused by insufficient autophagy in foam cells is likely due to the shutdown of the antioxidant effect of Nrf2 and the enhancement of the negative effect of Nrf2 caused by insufficient autophagy. Therefore, the positive feedback effect of insufficient intracellular autophagy and Nrf2 deposition is detrimental to cell survival and is a vicious cycle process. However, how the negative effect generated by Nrf2 drives foam cells toward death is a question we continue to explore in the following.

Insufficient autophagy of foam cells initiates the negative effect of Nrf2 and accelerates the death of foam cells. a THP-1 macrophages were treated with 50 µg/ml ox-LDL in 6-well plates for 48 h, followed by 30 µM CQ for 12 h and then t-BHQ or ML385 for 12 h. The expression of Nrf2 and p62 in the cells was detected by WB. b–c Nrf2 and p62 expression levels were calculated for each band using Image J analysis, each data set is from three independent tests and is expressed as a mean value. d THP-1 cells were added to a 96-well plate and induced into macrophages with PMA, and then the cells were divided into 5 groups, and each experiment was repeated three times. The first group was a blank control group. The second group was treated with 50 µg/ml ox-LDL for 48 h after 24 h of culture, and the third group was treated with 50 µg/ml ox-LDL for 48 h after 12 h of culture, followed by 30 µM CQ for 12 h. The fourth group was treated with 50 µg/ml ox-LDL for 48 h, followed by 30 µM CQ for 12 h, and then t-BHQ for 12 h. The fifth group was treated with 50 µg/ml ox-LDL for 48 h, 30 µM CQ for 12 h, and then ML385 for 12 h. During this period, the culture medium was changed every 12 h with 2% FBS RPMI1640. After treatment, CCK8 reagent was added and cell activity was detected by enzyme marker. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns indicates non-significant between specific designated groups
Insufficient autophagy in foam cells initiates Nrf2-induced ROS accumulation
With further probing, it was found that intracellular ROS levels were significantly elevated when autophagy was deficient in foam cells, and this intracellular ROS accumulation became more pronounced with the addition of the Nrf2 promoter t-BHQ, which in turn decreased intracellular ROS levels with the addition of ML385, an inhibitor of Nrf2 (Fig. 4a, b). This result suggests that the negative effect of Nrf2 initiated by insufficient autophagy in foam cells may be shaped by Nrf2-induced ROS accumulation, and the accumulation of intracellular ROS leads the cells toward death.

Insufficient autophagy of foam cells initiates Nrf2-induced ROS accumulation. a THP-1 cells were added to a 6-well plate and induced into macrophages with PMA, and then the cells were divided into 4 groups. The first group was treated with 50 µg/ml ox-LDL for 48 h after 24 h of culture, and the second group was treated with 50 µg/ml ox-LDL for 48 h after 12 h of culture, followed by 30 µM CQ for 12 h. The third group was treated with 50 µg/ml ox-LDL for 48 h, followed by 30 µM CQ for 12 h, and t-BHQ for 12 h. The fourth group was treated with 50 µg/ml ox-LDL for 48 h, 30 µM CQ for 12 h, and then ML385 for 12 h. The cells were changed every 12 h in 2% FBS RPMI1640 culture medium. After treatment the cells were digested down with trypsin, loaded with a good fluorescent probe DCFH-DA, and the intracellular ROS levels were observed by fluorescence microscopy. b The intracellular ROS levels were quantified by FCM
The negative effect of Nrf2 further promotes ferroptosis in foam cells
Interestingly, we further found that the reduced cell survival due to insufficient autophagy in foam cells could be reversed by the ferroptosis inhibitor Liproxstatin-1 (Fig. 5a). Moreover, the expression of ferroptosis-associated protein GPX4 was significantly reduced when foam cell autophagy was insufficient, and the addition of ferroptosis inhibitor Liproxstatin-1 reversed this phenomenon (Fig. 5b, d). In contrast, treatment of cells with the ferroptosis inhibitor Liproxstatin-1 failed to affect the intracellular Nrf2 expression level (Fig. 5b, c). Whereas the addition of t-BHQ, a promoter of Nrf2 in the presence of autophagy inhibition in foam cells, revealed that the expression of intracellular ferroptosis-related proteins GPX4 and xCT was decreased, the addition of ML385, an inhibitor of Nrf2, revealed that the expression of intracellular ferroptosis-related proteins GPX4 and xCT was increased and was higher than that in the group of cells with only CQ (Fig. 6a, c, d). The above results suggest that the negative effect of Nrf2 may promote ferroptosis in foam cells, and the specific mechanism may be related to the accumulation of intracellular ROS caused by Nrf2.

Reduced cell survival and reduced GPX4 expression due to insufficient autophagy in foam cells can be reversed by Liproxstatin-1. a THP-1 cells were added to a 96-well plate and induced into macrophages with PMA, and then the cells were divided into 4 groups, and each experiment was repeated three times. The first group was the control group. The second group was treated with 50 µg/ml ox-LDL for 48 h after 12 h of culture. The third group was treated with 50 µg/ml ox-LDL for 48 h followed by 30 µM CQ for 12 h. The fourth group was treated with 50 µg/ml ox-LDL for 48 h followed by 30 µM CQ and 10 µM Liproxstatin-1 for 12 h. During this period. The cells were changed every 12 h in 2% FBS RPMI1640 culture medium. After treatment, CCK8 reagent was added and cell activity was detected by enzyme marker. b The above experiments were repeated in 6-well plates, and intracellular Nrf2 and GPX4 expression was detected by WB. c and d The expression levels of Nrf2 and GPX4 were calculated by analyzing each band using Image J, with each data set from three independent tests and expressed as mean values. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns indicates non-significant between specific designated groups

The negative effect of Nrf2 reduces the expression of ferroptosis-related proteins GPX4 and xCT. a THP-1 cells were added to 6-well plates and induced into macrophages with PMA, and then the cells were divided into 4 groups. The first group was treated with 50 µg/ml ox-LDL for 48 h after 24 h of culture, and the second group was treated with 50 µg/ml ox-LDL for 48 h after 12 h of culture, followed by 30 µM CQ for 12 h. The third group was treated with 50 µg/ml ox-LDL for 48 h, followed by 30 µM CQ for 12 h, and t-BHQ for 12 h. The fourth group was treated with 50 µg/ml ox-LDL for 48 h, 30 µM CQ for 12 h, and then ML385 for 12 h. The culture medium was changed every 12 h with 2% FBS RPMI1640. The expression of intracellular Nrf2, xCT and GPX4 was detected by WB after treatment. b–d The expression levels of Nrf2, xCT and GPX4 were calculated using Image J analysis for each band (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns indicates that the specific designated groups not significant between), with each set of data from three independent tests and expressed as mean values
Discussion
We explored the relationship between autophagy and ferroptosis in AS by constructing a human foam cell model and found that deficient autophagy in late stage AS foam cells can lead to abnormal accumulation of Nrf2. Deficient autophagy in AS may initiate a pathogenic gene program that manipulates Nrf2 to downregulate Nrf2-mediated defense through the accumulation of p62 [19]. Abnormal functioning Nrf2 can lead to the accumulation of intracellular ROS and even further promote the development of cellular ferroptosis.
The present experimental study overturned the inherent view that Nrf2 only facilitates cell survival such as anti-oxidative stress and promoting iron metabolism [20]. It shows that Nrf2 also has a dual role in the general environment of the organism, and the abnormal accumulation and activation of Nrf2 promotes the development of AS. However, the mechanism by which Nrf2 promotes cellular ferroptosis in the presence of aberrant activation remains to be further explored. Although Basit et al. [21]showed that the accumulation of intracellular ROS is one of the important mechanisms of ferroptosis formation, and the present experiment also confirmed that activation of Nrf2 in the presence of insufficient autophagy leads to the accumulation of intracellular ROS, this is not sufficient to elucidate the specific mechanism by which Nrf2 promotes ferroptosis. It has been shown that iron overload can lead to damage of vascular endothelial cells by reactive oxygen ROS and lipid peroxidation, and that downregulation of the ferroptosis-related protein GPX4 and upregulation of ACSL4 caused by the ferroptosis inducer Erastin exacerbate the development of AS by causing ferroptosis in endothelial cells [22]. In addition, iron overload can aggravate AS plaque formation and instability by promoting foam cell formation and inflammatory response in AS and damaging smooth muscle cells [23]. It has been found that in oxidized low-density lipoprotein (OX-LDL)-treated mouse aortic endothelial cells, the ferroptosis inhibitor ferrostatin-1 inhibited iron overload state and lipid peroxidation, and enhanced the activity of xCT and GPX4, improving cell viability and reducing cell death [24]. However, the iron chelator desferricoprogen (DFC) inhibited plaque lipid oxidation and reduced plaque formation in APOE−/− mice on an AS-causing diet [25]. All of the above experimental studies illustrate the important role of iron overload in promoting the AS process. And Nrf2 can cause ferroptosis in cardiomyocytes of mice with cardiomyopathy by mediating the upregulation of heme oxygenase-1 (Hmox1), which results in accelerated heme degradation and systemic non-heme iron accumulation [26]. Therefore, whether the aberrant accumulation and activation of Nrf2 can regulate ferroptosis through its regulation of intracellular iron metabolism is a question that deserves further investigation.
Abbreviations
- AS:
-
Atherosclerosis
- AP:
-
Atherosclerotic plaque
- ATG5:
-
Autophagy-related gene5
- ATG7:
-
Autophagy-related gene7
- CQ:
-
Chloroquine
- CCK8:
-
Cell Counting Kit-8
- DFC:
-
Iron chelator desferricoprogen
- FCM:
-
Flow cytometry analysis
- FBS:
-
Fetal bovine serum
- Hmox1:
-
Heme oxygenase-1
- GPX4:
-
Glutathione Peroxidase 4
- LC3:
-
Microtubule-associated protein 1 light chain 3
- Lipro-1:
-
Liproxstatin-1
- Nrf2:
-
Nuclear factor erytheroid-derived-2-like 2
- ox-LDL:
-
Oxidized-low-density lipoprotein
- p62/SQSTM1:
-
Sequestosome 1
- PMA:
-
Phorbol-12-myristate-13-ace-tate
- ROS:
-
Reactive oxygen species
- tBHQ:
-
Tert-butylhydroquinone
- Xct:
-
Cystine-glutamic acid reverse transporter light chain protein
- WB:
-
Western blot
References
Moore KJ, Tabas I (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145(3):341–355. https://doi.org/10.1016/j.cell.2011.04.005
Martinet W, Coornaert I, Puylaert P, De Meyer GRY (2019) Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol 10(5):306–312. https://doi.org/10.3389/fphar.2019.00306
Zhou Y, Zhou H, Hua L, Hou C, Jia Q, Chen J, Zhang S, Wang Y, He S, Jia E (2021) Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med 171(2):55–68. https://doi.org/10.1016/j.freeradbiomed.2021.05.009
Evans TD, Jeong SJ, Zhang X, Sergin I, Razani B (2018) TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 14(4):724–726. https://doi.org/10.1080/15548627.2018.1434373
Kumar S, Nanduri R, Bhagyaraj E, Kalra R, Ahuja N, Chacko AP, Tiwari D, Sethi K, Saini A, Chandra V, Jain M, Gupta S, Bhatt D, Gupta P (2020) Vitamin D3-VDR-PTPN6 axis mediated autophagy contributes to the inhibition of macrophage foam cell formation. Autophagy 2020:1–17. https://doi.org/10.1080/15548627.2020.1822088
Osonoi Y, Mita T, Azuma K, Nakajima K, Masuyama A, Goto H, Nishida Y, Miyatsuka T, Fujitani Y, Koike M, Mitsumata M, Watada H (2018) Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 14(11):1991–2006. https://doi.org/10.1080/15548627.2018.1501132
Violi F, Carnevale R, Loffredo L, Pignatelli P, Gallin JI (2017) NADPH oxidase-2 and atherothrombosis: insight from chronic granulomatous disease. Arterioscler Thromb Vasc Biol 37(2):218–225. https://doi.org/10.1161/ATVBAHA.116.308351
Zhang J, Ma CR, Hua YQ, Li L, Ni JY, Huang YT, Duncan SE, Li S, Gao S, Fan GW (2021) Contradictory regulation of macrophages on atherosclerosis based on polarization, death and autophagy. Life Sci 276(6):118957. https://doi.org/10.1016/j.lfs.2020.118957
Chen Y, Li N, Wang H, Wang N, Peng H, Wang J, Li Y, Liu M, Li H, Zhang Y, Wang Z (2020) Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci 247(6):117–125. https://doi.org/10.1016/j.lfs.2020.117425
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, Wang J (2019) Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med 23(8):4900–4912. https://doi.org/10.1111/jcmm.14511
Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, Wang X, Cui T (2020) Autophagy Inhibition Enables Nrf2 to Exaggerate the Progression of Diabetic Cardiomyopathy in Mice. Diabetes 69(12):2720–2734. https://doi.org/10.2337/db19-1176
Zhu Y, Zhang Y, Huang X, Xie Y, Qu Y, Long H, Gu N, Jiang W (2019) Z-Ligustilide protects vascular endothelial cells from oxidative stress and rescues high fat diet-induced atherosclerosis by activating multiple NRF2 downstream genes. Atherosclerosis 284(21):110–120. https://doi.org/10.1016/j.atherosclerosis.2019.02.010
Mimura J, Itoh K (2015) Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic Biol Med 88(Pt B):221–232. https://doi.org/10.1016/j.freeradbiomed.2015.06.019
Hashimoto K, Simmons AN, Kajino-Sakamoto R, Tsuji Y, Ninomiya-Tsuji J (2016) TAK1 regulates the Nrf2 antioxidan system through modulating p62/SQSTM1. Antioxid Redox Signal 25(17):953–964. https://doi.org/10.1089/ars.2016.6663
Lazaro I, Lopez-Sanz L, Bernal S, Oguiza A, Recio C, Melgar A, Jimenez-Castilla L, Egido J, Madrigal-Matute J, Gomez-Guerrero C (2018) Nrf2 activation provides atheroprotection in diabetic mice through concerted upregulation of antioxidant, anti-inflammatory, and autophagy mechanisms. Front Pharmacol 9(9):819–823. https://doi.org/10.3389/fphar.2018.00819
Wu J, Ni Y, Yang Q, Mao J, Zhu X, Tao S, Kato K, Zhang J, Wang D, Yamanaka K, An Y (2020) Long-term arsenite exposure decreases autophagy by increased release of Nrf2 in transformed human keratinocytes. Sci Total Environ 734(16):139425. https://doi.org/10.1016/j.scitotenv.2020.139425
Patel KM, Strong A, Tohyama J, Jin X, Morales CR, Billheimer J, Millar J, Kruth H, Rader DJ (2015) Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ Res 116(5):789–796. https://doi.org/10.1161/CIRCRESAHA.116.305811
Liu Z, Zhu H, Dai X, Wang C, Ding Y, Song P, Zou MH (2017) Macrophage liver kinase B1 inhibits foam cell formation and atherosclerosis. Circ Res 121(9):1047–1057. https://doi.org/10.1161/CIRCRESAHA.117.311546
Liu J, Wang C, Li J, Yu Y, Liu Y, Liu H, Peng Q, Guan X (2021) Autophagy blockage promotes the pyroptosis of ox-LDL-treated macrophages by modulating the p62/Nrf2/ARE axis. J Physiol Biochem. https://doi.org/10.1007/s13105-021-00811-2
Yang K, Song H, Yin D (2021) PDSS2 inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J Cardiovasc Pharmacol 77(6):767–776. https://doi.org/10.1097/FJC.0000000000001030
Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S, Kopitz C, Heroult M, Hgm Willems P, Koopman WJ (2017) Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis 8(3):e2716. https://doi.org/10.1038/cddis.2017.133
Xiao FJ, Zhang D, Wu Y, Jia QH, Zhang L, Li YX, Yang YF, Wang H, Wu CT, Wang LS (2019) miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem Biophys Res Commun 515(3):448–454. https://doi.org/10.1016/j.bbrc.2019.05.147
Vinchi F, Porto G, Simmelbauer A, Altamura S, Passos ST, Garbowski M, Silva AMN, Spaich S, Seide SE, Sparla R, Hentze MW, Muckenthaler MU (2020) Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J 41(28):2681–2695. https://doi.org/10.1093/eurheartj/ehz112
Bai T, Li M, Liu Y, Qiao Z, Wang Z (2020) Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med 160(1):92–102. https://doi.org/10.1016/j.freeradbiomed.2020.07.026
Potor L, Eva SK, Hegedus H, Petho D, Szabo Z, Szigeti ZM, Pocsi I, Trencsenyi G, Szikra D, Garai I, Gall T, Combi Z, Kappelmayer J, Balla G, Balla J (2020) The fungal iron chelator desferricoprogen inhibits atherosclerotic plaque formation. Int J Mol Sci 21(13):4746–4749. https://doi.org/10.3390/ijms21134746
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu X, Gao F, Zhu N, Yin X, Chen Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA 116(7):2672–2680. https://doi.org/10.1073/pnas.1821022116
Funding
This study was supported by the grants from the National Natural Science Foundation of China (No. 81672084).
Author information
Authors and Affiliations
Contributions
First author: do experiments, write articles, participate in part of the experimental design. Second, third, fourth and fifth Author: assist in completing the experiment. Corresponding author: participate in article revision, experimental design and supervision, financial support. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Informed consent
All authors were informed and agreed to sign.
Research involving human participants and/or animals
This study does not involve human participants and/or animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peng, Q., Liu, H., Luo, Z. et al. Effect of autophagy on ferroptosis in foam cells via Nrf2. Mol Cell Biochem 477, 1597–1606 (2022). https://doi.org/10.1007/s11010-021-04347-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04347-3