
Abstract
Activating mutations within the tyrosine kinase (TK) domain of epidermal growth factor receptor (EGFR) gene are observed in 10 ~ 30% of the patients diagnosed with non-small cell lung cancer (NSCLC), and are causally related to NSCLC initiation and progression. Treatments with tyrosine kinase inhibitors (TKIs) targeting EGFR significantly improve the outcome of NSCLC patients with EGFR mutation, but are often associated with drug resistance, which is the main cause of treatment failure and cancer relapse. In the present study, by screening the transcriptome of NSCLC patients, we found that EGFR activation is highly correlated with the up-regulation of mitotic regulator, never in mitosis gene A-related kinase 2 (NEK2). NEK2 overexpression is associated with the poor survival of EGFR-mutant patients but not the wild-type patients. Further functional validation revealed that EGFR mutation induces NEK2 expression by activating ERK signaling pathway. Elevated NEK2 level promotes the rapid cell cycle progression and favors the rapid proliferation of EGFR-mutant NSCLC cells. Of note, NEK2 overexpression also impairs the efficacy of TKI treatment via inhibiting apoptosis, while depleting NEK2 suppresses cell growth and restored the sensitivity of TKI in NSCLC cells. Taken together, our study revealed that NEK2 is an oncogene regulated by EGFR mutation and is involved in disease progression and treatment response in NSCLC with EGFR mutation. These findings will pave the road for optimizing personalized treatment strategies to overcome drug resistance and improve the prognosis of lung cancer patients with EGFR mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer morbidity and mortality worldwide, and non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases [1]. Notably, there are 10–30% of patients with NSCLC bear activating mutations in the epidermal growth factor receptor (EGFR) gene [2,3,4]. The majority of the mutations (~ 90%) are characterized as short in-frame deletions in exon 19 (delE746-A750) and point mutation in exon 21 (Leu858Arg) within the tyrosine kinase (TK) domain, which lead to the constitutive activation of EGFR-mediated cell proliferation and cell survival and has been recognized as an important driver of lung tumorigenesis [5, 6].
Tyrosine kinase inhibitors (TKIs) of EGFR, such as gefitinib, erlotinib and afatinib, are capable of inhibiting the activation of mutant EGFR and have been used as the first line treatment for EGFR-mutant NSCLC at advanced disease stages. A large portion (60 ~ 85%) of the patients receiving EGFR-TKIs show favorable treatment response. The progression-free survival and overall survival in these patients are also largely improved compared to chemotherapy [7, 8]. Although growing evidence confirms the significant benefit of EGFR-targeted TKIs for lung cancer patients, most of the patients eventually develop drug resistance, which is the major cause of treatment failure [9]. It has been found that a secondary mutation in the EGFR gene (Thr790Met) causes conformational change to prevent TKI binding and is responsible for a large fraction (~ 50%) of drug resistance in EGFR-mutant patients [10, 11]. Other genetic variations, such as mutations in v-Raf murine sarcoma viral oncogene homolog B (BRAF) gene and phosphatidylinositol-4,5-bisphosphate 3-kinase (PIK3CA) gene, genetic amplifications of MNNG HOS transforming (MET) gene and human epidermal growth factor receptor 2 (HER2) gene, have also been frequently associated with EGFR-TKI resistance and are found to promote cancer progression via mechanisms different from EGFR activation [12,13,14]. Despite the great efforts devoted to investigating the underlying mechanism and developing novel methods to improve drug efficacy, the molecular process by which cancer cells acquire such resistance is yet to be investigated and disease relapse inevitably occurs in most patients over a period of 9 to 13 months. All these prior findings imply the complexity of NSCLC development in the setting of EGFR mutation. Therefore, understanding how EGFR mutations initiate tumor development and how these EGFR-mutant tumor cells persist as residual disease during treatment is highly required for the optimization of current treatment strategy and the development of additional, alternative approaches to treat EGFR inhibitor resistant cells.
Here we focus on the discovery of new mechanisms related to EGFR mutation and EGFR-TKI resistance in NSCLC. Combining transcriptomics-based screening and functional validation, we found that cell mitotic regulator, never in mitosis gene A-related kinase 2 (NEK2), is an oncogene specific to EGFR-mutant NSCLC, while knockdown of NEK2 suppresses disease progression and enhances the response to TKI treatment in EGFR-mutant NSCLC cells. Findings from our studies will hopefully lead to improved treatments that overcome resistance to EGFR inhibitors and increase the survival of lung cancer patients.
Methods
Cell culture
Cell lines used in the study include normal lung epithelial cells Beas-2B, EGFR wild-type NSCLC cells A549 and H1703 as well as EGFR-mutant NSCLC cells PC-9 and H3255. PC-9 cells have E746-A750 deletion in exon 19 (Del19) and H3255 cells bear Leu858Arg mutation in exon 21(L858R). All cell lines were obtained from Cell Resource Center of Shanghai Institutes for Biological Sciences. Gefitinib-resistant cell lines PC-9/GR and H3255/GR were established by continuously culturing parental PC-9 and H3255 cells in gefitinib-containing media [15]. The gefitinib concentration started from 0.01 μM and was gradually increased when the cells acquired resistance. BEAS-2B cell was cultured in DMEM media. PC-9, H3255, PC-9/GR and H3255/GR cells were growth in RPMI-1640 medium. All cell cultures were supplemented with 10% fetal bovine serum, 2 mM glutamine 1% penicillin and 1% streptomycin, and incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Stable cell line expressing mutant EGFR or NEK2
pBabe-puro vectors containing mutant EGFR (Del19 or L858R) and empty pBabe-Puro vector were transfected into 293T cells together with packaging plasmid using Lipofectamine 2000. Recombinant lentivirus was collected 2 days after transfection and was used to infect Beas-2B cells. Puromycin was used to select stable cells expressing mutant EGFR.
To overexpress NEK2 (NEK2 OE), recombinant lentivirus expressing either empty vector or pLVX-Puro containing the NEK2 cDNA sequence were obtained by co-transfecting 293T cells with packaging vector, and were transduced into PC-9 and H3255 cells. Transduction efficiency was evaluated using puromycin.
NEK2 knockdown
Small interfering RNA (siRNA) against NEK2 (siNEK2) were introduced into PC-9/GR and H3255/GR cells using Lipofectamine 2000 according to manufacturer’s instruction, followed by 72 h incubation at 37 °C. siRNA with nonsense sequence (siCtrl) was also used to transfect the cells and was used as negative control.
Transcriptome data and gene set enrichment analysis (GSEA)
Publicly available transcriptome data GSE31210 and GSE13213 were obtained from Gene Expression Omnibus (GEO) database. GSEA analysis was performed using data preranked based on the correlation of a gene with EGFR expression. Gene Ontology database was used. Gene sets with FDR < 0.05 were considered as significantly correlated with EGFR.
Quantitative polymerase chain reaction (qPCR)
Trizol reagent was used to extract total RNA from Beas-2B and NSCLC cells treated under different conditions. cDNA was synthesized from extract RNA via reverse transcription and was used as the template for qPCT assay. qPCR was performed in triplicate using gene specific primers, including NEK2: forward 5′-ACGGAAGTTCCTGTCTCTGGCA-3′; reverse 5′-GCACTTGGACTTAGATGTGAGCT-3′; GAPDH: forward 5′-GTCTCCTCTGACTTCAACAGCG-3′; reverse 5′-ACCACCCTGTTGCTGTAGCCAA-3′. The relative NEK2 mRNA level was calculated using 2−ΔΔCT method with GAPDH as the reference.
Cell viability assay
Cell viability was assessed using MTS assay. To measure cell proliferation, NSCLC cells were plated in 96-well plate and cell viability was assessed every 12 h for up to 96 h. To evaluate drug sensitivity, NSCLC cells were plated in 96-well plate for 12 h and were then treated with different concentrations of TKIs, including gefitinib, erlotinib and afatinib. Cell viability was assessed after incubation for 72 h. All the experiment was performed with three replications.
Cell cycle assay
NSCLC cells overexpressing NEK2 or empty vector were harvested. After ethanol fixation and permeabilization, the cells were stained with propidium iodide (PI) in the presence of RNAse A and were analyzed by FACSCalibur flow cytometry. Experiment was performed in triplicate.
Carboxyfluorescein succinimidyl ester (CFSE)
CFSE enables long last fluorescent labeling of live cells. The fluorescent signal decreases as cells divide, allowing for the monitoring of cell proliferation. On Day 0, suspended NSCLC cells were labeled with CFSE reagent (BD bioscience) following manufacturer’s instruction, and were plated in 6-well plate. After 48 h incubation, the cells were harvested and cell proliferation was assessed by flow cytometry. The experiment was replicated three times.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
TUNEL assay was used to detect apoptotic cells. NSCLC cells were treated with 0.1 µM gefitinib for 48 h according to previous studies [16, 17]. After formaldehyde fixation, cells were stained with TUNEL labeling reagent per manufacturer’s instruction. Nuclei were counterstained with DAPI. TUNEL and DAPI positive cells were visulized under fluorescence microscopy and photographed.
Apoptosis by flow cytometry
Cell apoptosis was also assessed by Annexin V/PI double staining method. NSCLC cells were plated in 6-well plates for 12 h and were treated with 0.1 µM gefitinib for another 48 h [16, 17]. After that, cells were harvested and labeled with Annexin V and PI. The fluorescent signal was detected using flow cytometry.
Western blot assay
NSCLC cells were lysed using RIPA lysis buffer and were centrifuged at 15,000×g for 30 min. The supernatant was collected and the total protein concentration was quantified using BCA method. Equal amount of proteins from each sample were separated by denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membrane (Millipore). After blocking with 5% non-fat skim milk for 1 h, the membrane were incubated overnight with primary antibodies NEK2, pEGFR, EGFR, pERK, ERK, pAKT, AKT, pCDC20, CDC20, Ki-67, Bax, Caspase 3 and GAPDH. The protein signal was detected using secondary antibody conjugated with horseradish peroxidase and visualized using enhanced chemiluminescence detection system (PerkinElmer).
Results
NEK2 is highly associated with EGFR activation
As EGFR mutations cause constitutive EGFR activation even in the absence of extracellular ligand, we thus screened for the biological functions and genes most correlated with EGFR expression using public transcriptome datasets GSE31210 and GSE13213, which comprise gene expression data obtained from 226 and 117 NSCLC patients, respectively. All the genes were ranked according to their correlation coefficients with EGFR expression and were subjected to gene set enrichment analysis (GSEA). As a result, chromosome segregation, an essential part of cell division, was revealed to be most associated with EGFR activation (Fig. 1a). Moreover, by analyzing the top genes involved chromosome segregation, we found that never in mitosis gene A-related kinase 2 (NEK2) was highly correlated with EGFR in both patient cohorts (Fig. 1b), indicating that NEK2 might be a common responder of EGFR activation in NSCLC patients.

Transcriptome screening for EGFR-related biological functions. a The top biological functions associated with EGFR expression in two different NSCLC transcriptome datasets GSE31210 and GSE13213. Data are ranked according to normalized enrichment score (NES) obtained from GSEA analysis. b Chromosome segregation was positively correlated with EGFR and NEK2 was identified as a potential candidate for further investigation
NEK2 is an oncogene specific to EGFR mutation
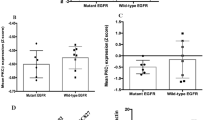
NEK2 is an oncogene overexpressed in a wide range of human cancers and predictive of poor prognosis [18,19,20,21,22]. As shown in Fig. 2a, NEK2 was significantly correlated with EGFR expression. Patients with high EGFR expression show elevated NEK2 levels than those with low EGFR expression (Fig. 2b). Of note, when we stratified the NSCLC patients based on their EGFR mutation state, this kind of correlation was only evident in patients with EGFR mutation but not in patients with wild-type EGFR (Fig. 2a, c). Moreover, in EGFR-mutant NSCLC, NEK2 expression was also correlated with patient prognosis. Patients with high NEK2 levels were at largely increased risk of death within 10 years after diagnosis (Fig. 2d). However, we didn’t see a statistically significant association between NEK2 and the survival of EGFR wild-type patients (Fig. 2d). A significant difference was observed between EGFR-mutant and wild-type NSCLC when combining the two datasets (GSE31210, GSE13213) using meta-analysis (Fig. 2e). All these findings suggested that the oncogenic function exerted by NEK2 is more pronounced in EGFR-mutant NSCLC.

NEK2 is an oncogene specific to EGFR mutation. a The correlation of NEK2 expression with EGFR expression in NSCLC patients with mutant EGFR (EGFR Mut) and wild-type EGFR (EGFR WT) in two different transcriptome datasets GSE31210 and GSE13213. b NEK2 levels in patients with high and low EGFR expression. Patients with EGFR levels higher than average are classified into high EGFR group, and those with EGFR levels lower than average are in low EGFR group. c The correlation coefficient of NEK2 and EGFR expression in NSCLC patients with and without EGFR mutation. d The association of NEK2 expression with the overall survival of NSCLC patient with mutant and wild-type EGFR. e The risk of lung cancer death regarding NEK2 expression was assessed using Cox proportional-hazards model for EGFR-mutant NSCLC patients and EGFR wild-type NSCLC patients, respectively, and was compared between the two patient groups. Meta-analysis was performed to combine the results from GSE31210 and GSE13213 datasets. f NEK2 expression in public transcriptome dataset (GSE75309) evaluating gene expression change in response to gefitinib treatment. g NEK2 expression in public transcriptome dataset (GSE112274) assessing gene expression change during the development of TKI resistance. *p < 0.05; **p < 0.01; ***p < 0.001
Of note, by analyzing the transcriptome data obtained from EGFR-mutant NSCLC cells (GSE75309, GSE112274), we found that tyrosine kinase inhibitors (TKIs) treatment dramatically inhibited NEK2 expression (Fig. 2f) and NEK2 expression gradually increased with the generation of TKI resistance (Fig. 2g), suggesting that NEK2 is playing a role in the response to EGFR-targeted treatment.
EGFR mutation induces NEK2 expression via ERK signaling pathway
The above transcriptome data mining identified NEK2 as a potential regulator in EGFR-mutant NSCLC, we then specifically investigated NEK2 function in lung cells expressing activating EGFR mutation. As exon 19 E746-A750 deletion (Del19) and exon 21 Leu858Arg mutation (L858R) account for up to 90% of EGFR mutations in NSCLC, we first constructed cells line expressing those mutations in normal lung epithelial cells Beas-2B, and we found that introducing EGFR mutations increased NEK2 expression in both mRNA and protein levels (Fig. 3a, b). Moreover, in NSCLC cells, those with Del19 or L858R mutations (PC-9, H3255) showed higher NEK2 mRNA and protein expression than the cells with wild-type EGFR (A549, H1703) (Fig. 3c, d). As EGFR mutation results in consistent EGFR activation, these findings further supporting a correlation between EGFR activation and NEK2 overexpression, while inhibiting EGFR with gefitinib significantly suppressed NEK2 expression in NSCLC cells with EGFR mutation (PC-9, H3255) (Fig. 3e, f).

NEK2 is induced by EGFR mutation via ERK signaling pathway. a EGFR mutation was introduced into Beas-2B cells, which originally expresses wild-type EGFR, and the mRNA level of NEK2 was measured. b NEK2 expression at protein level was measured in Beas-2B cells with and without EGFR mutations. c NEK2 mRNA expression in NSCLC cells with wild-type and mutant EGFR. d NEK2 protein expression in NSCLC cells with wild-type and mutant EGFR. e The expression status of NEK2 and the activation status of EGFR, ERK and AKT was measured in EGFR-mutant NSCLC cells PC-9 (Del19) and H3255 (L858R) under different treatment condition. SCH772984 is ERK inhibitor and LY294002 is AKT inhibitor. f The mRNA level of NEK2 in EGFR-mutant NSCLC cells under different treatment condition. *p < 0.05; **p < 0.01; ***p < 0.001
EGFR mutation has been reported to activate ERK and AKT signaling pathways [23]. Consistent with this, decreased phosphorylation of ERK and AKT was observed following gefitinib treatment, suggesting the deactivation of the downstream oncogenic pathways (Fig. 3e). Moreover, specifically suppressing ERK signaling pathway with ERK inhibitor (SCH772984) also reduced NEK2 expression in EGFR-mutant NSCLC cells, while AKT inhibitor (LY294002) showed no influence on NEK2 level (Fig. 3e, f). We thus concluded that EGFR mutation induced NEK2 overexpression in NSCLC cells is mediated by ERK signaling pathway but not AKT signaling pathway.
NEK2 overexpression promotes the proliferation of EGFR-mutant NSCLC cells
To understand the function of NEK2 in NSCLC with EGFR mutation, we overexpressed NEK2 in EGFR-mutant NSCLC cell lines PC-9 and H3255 (NEK2 OE). NEK2 is a mitotic regulator involved in centrosome separation, chromosome condensation and spindle formation. Elevated NEK2 may thus favor the progression of cell division. In our study, NEK2 overexpression in EGFR-mutant NSCLC cells was associated with coordinated activation of cell division cycle protein 20 (CDC20), a cell division regulator initiating chromosome separation and mitotic exit (Fig. 4a). A decrease in cell proportion arrested at G2/M phase was also observed following NEK2 overexpression (Fig. 4b, c), consistent with accelerated cell division process. Rapid cell cycle progression is coupled with rapid cell proliferation. As evidenced by more diluted CFSE signal (Fig. 4d, e) and faster growth rate (Fig. 4f), the proliferation of EGFR-mutant cells was enhanced by NEK2 overexpression. This result was also supported by the up-regulation of proliferation marker Ki-67 in NEK2-overexpressed cells (Fig. 4a). Taken together NEK2 facilitates the rapid division and growth of EGFR-mutant NSCLC cells.

NEK2 overexpression promotes the proliferation of EGFR-mutant NSCLC cells. a The expression of cell division regulator CDC20 and cell proliferation marker Ki-67 in EGFR-mutant NSCLC cells PC-9 (Del19) and H3255 (L858R) expressing empty vector and overexpressing NEK2 (NEK2 OE). b, c The changes of cell cycle distribution in EGFR-mutant NSCLC cells with and without NEK2 overexpression. d The influence of NEK2 overexpression on the proliferation of EGFR-mutant NSCLC cells was assessed using CFSE method. e Comparison of CFSE signal in EGFR-mutant NSCLC cells with and without NEK2 overexpression. f The influence of NEK2 on the viability of EGFR-mutant NSCLC cells after different incubation periods. *p < 0.05; **p < 0.01; ***p < 0.001
NEK2 overexpression impairs the efficacy of TKI treatment
As EGFR mutation increases the sensitivity to TKI treatment, we then tested the influence of NEK2 on the response of EGFR-mutant cells to several TKIs, including gefitinib, erlotinib and afatinib. As revealed by drug sensitivity assay (Fig. 5a), NEK2 promoted cell viability in the presence of all these TKIs. More drugs were required by NEK2-overexpressed cells to inhibit 50% cell growth (IC50) (Fig. 5b). As gefitinib is the first EGFR inhibitor approved for NSCLC treatment, we then specifically investigate the influence of NEK2 on gefitinib treatment. As shown in Fig. 5c and d, when exposed to gefitinib, cells overexpressing NEK2 were detected with less TUNEL positive staining. The expression level of pro-apoptotic Bax and the activity of caspase 3 were also decreased with NEK2 overexpression (Fig. 5e), suggesting impaired apoptosis on gefitinib treatment. All these results implied a role of NEK2 in TKI response, which might be associated with EGFR-TKI resistance, a major obstacle in clinical management of NSCLC.

NEK2 overexpression confers drug resistance in EGFR-mutant NSCLC cells. a The influence of NEK2 overexpression on gefitinib, erlotinib and afatinib response. b IC50 concentration of gefitinib, erlotinib and afatinib in EGFR-mutant NSCLC cells with and without NEK2 overexpression. c TUNEL assay of the apoptosis of EGFR-mutant NSCLC cells expressing empty vector (Vector) and overexpressing NEK2 (NEK2 OE). Cells were treated with 0.1 µM gefitinib for another 48 h. Scale bar: 50 µm. d Comparison of TUNEL positive cells between EGFR-mutant NSCLC cells with and without NEK2 overexpression. e The expression of apoptosis marker Bax and Caspase 3 was assessed in EGFR-mutant NSCLC cells. *p < 0.05; **p < 0.01; ***p < 0.001
NEK2 knockdown inhibit cell proliferation and restores drug sensitivity
To further investigate the function of NEK2 in EGFR-TKI resistance, we established gefitinib resistant cell lines (PC-9/GR and H3255/GR) from parental PC-9 and H3255 cells by long-term exposure to gefitinib. Compared to parental cells, gefitinib resistant cells were observed with increased NEK2 expression (Fig. 6a), which is consistent with our analysis of public transcriptome data in Fig. 2f.

NEK2 knockdown restores drug sensitivity in EGFR-mutant NSCLC cells. a NEK2 expression in EGFR-mutant NSCLC cells sensitive and resistant to gefitinib. b The influence of NEK2 knockdown on the expression and activity of cell division regulator CDC20, cell proliferation marker Ki-67 and apoptosis marker Bax and Caspase 3 in gefitinib resistant cells. c Cell proliferation assay in gefitinib resistant NSCLC cells with and without NEK2 depletion. d The influence of NEK2 knockdown on cellular response to gefitinib treatment in gefitinib resistant NSCLC cells. e Comparison the percentage of apoptotic cells between gefitinib resistant cells with and without NEK2 knockdown. Cells were treated with 0.1 µM gefitinib for another 48 h. f Drug sensitivity assay in gefitinib resistant cells. g Comparison of gefitinib IC50 concentration between gefitinib resistant cells with and without NEK2 knockdown. *p < 0.05; **p < 0.01; ***p < 0.001
As revealed by above results, NEK2 is involved in the proliferation of EGFR-mutant NSCLC cells, we then evaluated the influence of NEK2 on the growth of gefitinib resistant cells by depleting NEK2 expression using small interfering RNA (siRNA). As shown in Fig. 6b, NEK2-depleted cells were seen with deactivation of mitotic regulator CDC20, which may subsequently slow down cell cycle progression. In addition, decreased expression of proliferation marker Ki-67 as well as decreased proliferation rate were also observed in gefitinib resistant cells following NEK2 knockdown (Fig. 6b, c), indicating that NEK2 is responsible for the rapid growth of EGFR-TKI resistant cells. More importantly, as evidenced by increased level of Bax and active Caspase 3 (Fig. 6b), knockdown of NEK2 increased the apoptotic potential in gefitinib resistant cells. More NEK2-depleted cells were undergoing apoptotic cell death on gefitinib treatment (Fig. 6d, e). The cellular sensitivity to gefitinib was largely restored in those cells and the gefitinib IC50 concentration was significantly decreased (Fig. 6f, g), suggesting that NEK2 depletion increased TKI efficacy in EGFR-mutant NSCLC cells.
Discussion
Activating EGFR mutation is associated with an important subgroup of NSCLC patients [2,3,4]. The clinical application of EGFR-TKIs has dramatically improved the prognosis of NSCLC patients with EGFR mutation and is a milestone in targeted therapy of NSCLC [7, 8]. However, during the treatment process, most of the patients inevitably develop acquired resistance to TKIs, leading to treatment failure and disease relapse [9]. Given these, understanding the molecular processes related to EGFR mutation and EGFR-TKI resistance will provide valuable information to optimize NSCLC treatment and improve drug efficacy. In the present study, we provided evidence that activating EGFR mutation induces the expression of mitotic regulator NEK2 via ERK signaling pathway. NEK2 overexpression promotes the progression of EGFR-mutant NSCLC, impairs thus response to TKI treatment and is associated with the poor prognosis of EGFR-mutant NSCLC patients. More importantly, our analysis also revealed that the oncogenic function of NEK2 is more specific to EGFR mutation rather than wild type, highlighting the potential of NEK2 as a therapeutic target in this specific patient group.
NEK2 has been increasingly recognized as an oncogene associated with the initiation and progression of various human cancers, such as breast cancer, colorectal cancer, prostate cancer and lung cancer [18,19,20,21,22]. In our study, we found that NEK2 favors the NSCLC progression and drug resistance by promoting cell division and inhibiting apoptosis. In addition to this, NEK2 is also implicated in the transformation of indolent follicular lymphoma into aggressive diffuse large B-cell lymphoma [24, 25]. In ovarian cancer, NEK2 overexpression was reported to impair drug efficacy by activating drug efflux [26]. There is also evidence revealing that NEK2 induces cell metastasis by cooperating with other oncogenic Ras and Src signaling pathways, and this kind of cooperation is modulated by PI3K/Akt pathways [27]. More recently, NEK2 has been characterized with a role in autophagy, which is tightly associated with the response to various cancer treatments and may serve as a promising therapeutic target [28,29,30]. Moreover, many studies have identified a direct interaction of NEK2 with oncogene β-catenin [31, 32], implying the possible involvement of NEK2 in β-catenin-mediated oncogenic signaling network, such as Wnt signaling pathway and cell–cell adhesion. All these findings indicate NEK2 as a multifunctional oncogene, and the tumorigenic function of NEK2 in NSCLC as well as other types of cancer is thus far more complex than previously thought, which deserves to be investigated in the future studies.
EGFR mutation is now widely accepted as an important driver of lung cancer. It has been frequently reported that EGFR mutation initiates the growth, differentiation and angiogenesis of cancer cell via various oncogenic mechanisms [23]. For example, EGFR mutation has been reported to enhances the expression Cadherin-5 and promote angiogenesis by activating AKT signaling pathway [33]. EGFR activation also up-regulates PD-L1 through ERK/c-Jun axis and mediates immune escape in EGFR-mutant NSCLC [34]. Our study identified NEK2-mediated cell mitosis as a regulator of NSCLC progression specific to EGFR mutation. Moreover, according to our discovery transcriptome analysis, many other mitotic regulators and many other cellular functions, such as DNA replication, protein synthesis and mitochondrial oxidative phosphorylation, are also highly correlated with EGFR and are thus very likely to play a role in EGFR-mutant NSCLC. All these pieces of evidences imply a complex functional network regulated by EGFR mutation, which are closely related to various tumor phenotypes and affect disease development and treatment response. Therefore, systematically understanding the molecular alterations induced by EGFR mutation will provide novel insights to optimize the treatment and improve the survival of NSCLC patients.
Taken together, combining large-scale data mining and experimental validation, our study revealed that NEK2 is an oncogene highly correlated with EGFR mutation and is responsible for the progression of EGFR-mutant NSCLC, while depleting NEK2 expression suppresses the rapid growth of cancer cell and improves the efficacy of EGFR-TKIs, which could become a novel therapeutic option for lung cancer management in clinic.
References
Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL, Siegel RL (2019) Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 69:363–385. https://doi.org/10.3322/caac.21565
Sharma SV, Bell DW, Settleman J, Haber DA (2007) Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7:169–181. https://doi.org/10.1038/nrc2088
Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T (2004) Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 64:8919–8923. https://doi.org/10.1158/0008-5472.CAN-04-2818
Cancer Genome Atlas Research N (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543–550. https://doi.org/10.1038/nature13385
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139. https://doi.org/10.1056/NEJMoa040938
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500. https://doi.org/10.1126/science.1099314
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361:947–957. https://doi.org/10.1056/NEJMoa0810699
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y, Geater SL (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15:213–222. https://doi.org/10.1016/S1470-2045(13)70604-1
Wu SG, Shih JY (2018) Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer 17:38. https://doi.org/10.1186/s12943-018-0777-1
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792. https://doi.org/10.1056/NEJMoa044238
Wu SG, Liu YN, Tsai MF, Chang YL, Yu CJ, Yang PC, Yang JC, Wen YF, Shih JY (2016) The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 7:12404–12413. https://doi.org/10.18632/oncotarget.7189
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043. https://doi.org/10.1126/science.1141478
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:7526. https://doi.org/10.1126/scitranslmed.3002003
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44:852–860. https://doi.org/10.1038/ng.2330
Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S, Satomi R, Naoki K, Betsuyaku T (2013) Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res 11:759–767. https://doi.org/10.1158/1541-7786.MCR-12-0652
Li H, Zhou S, Li X, Wang D, Wang Y, Zhou C, Schmid-Bindert G (2013) Gefitinib-resistance is related to BIM expression in non-small cell lung cancer cell lines. Cancer Biother Radiopharm 28:115–123. https://doi.org/10.1089/cbr.2012.1268
Walsh AM, Lazzara MJ (2013) Regulation of EGFR trafficking and cell signaling by Sprouty2 and MIG6 in lung cancer cells. J Cell Sci 126:4339–4348. https://doi.org/10.1242/jcs.123208
Fang Y, Zhang X (2016) Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 15:895–907. https://doi.org/10.1080/15384101.2016.1152430
Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM (2004) The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res 64:7370–7376. https://doi.org/10.1158/0008-5472.CAN-04-0960
Takahashi Y, Iwaya T, Sawada G, Kurashige J, Matsumura T, Uchi R, Ueo H, Takano Y, Eguchi H, Sudo T, Sugimachi K, Yamamoto H, Doki Y, Mori M, Mimori K (2014) Up-regulation of NEK2 by microRNA-128 methylation is associated with poor prognosis in colorectal cancer. Ann Surg Oncol 21:205–212. https://doi.org/10.1245/s10434-013-3264-3
Zeng YR, Han ZD, Wang C, Cai C, Huang YQ, Luo HW, Liu ZZ, Zhuo YJ, Dai QS, Zhao HB, Liang YX, Zhong WD (2015) Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol 15:90. https://doi.org/10.1186/s12894-015-0085-7
Zhong X, Guan X, Dong Q, Yang S, Liu W, Zhang L (2014) Examining Nek2 as a better proliferation marker in non-small cell lung cancer prognosis. Tumour Biol 35:7155–7162. https://doi.org/10.1007/s13277-014-1935-8
da Cunha SG, Shepherd FA, Tsao MS (2011) EGFR mutations and lung cancer. Annu Rev Pathol 6:49–69. https://doi.org/10.1146/annurev-pathol-011110-130206
de Vos S, Hofmann WK, Grogan TM, Krug U, Schrage M, Miller TP, Braun JG, Wachsman W, Koeffler HP, Said JW (2003) Gene expression profile of serial samples of transformed B-cell lymphomas. Lab Invest 83:271–285. https://doi.org/10.1097/01.lab.0000053913.85892.e9
Andreasson U, Dictor M, Jerkeman M, Berglund M, Sundstrom C, Linderoth J, Rosenquist R, Borrebaeck CA, Ek S (2009) Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am J Hematol 84:803–808. https://doi.org/10.1002/ajh.21549
Liu X, Gao Y, Lu Y, Zhang J, Li L, Yin F (2014) Upregulation of NEK2 is associated with drug resistance in ovarian cancer. Oncol Rep 31:745–754. https://doi.org/10.3892/or.2013.2910
Das TK, Dana D, Paroly SS, Perumal SK, Singh S, Jhun H, Pendse J, Cagan RL, Talele TT, Kumar S (2013) Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis 2:e69. https://doi.org/10.1038/oncsis.2013.34
Xia J, He Y, Meng B, Chen S, Zhang J, Wu X, Zhu Y, Shen Y, Feng X, Guan Y, Kuang C, Guo J, Lei Q, Wu Y, An G, Li G, Qiu L, Zhan F, Zhou W (2020) NEK2 induces autophagy-mediated bortezomib resistance by stabilizing Beclin-1 in multiple myeloma. Mol Oncol 14:763–778. https://doi.org/10.1002/1878-0261.12641
Tang HW, Hu Y, Chen CL, Xia B, Zirin J, Yuan M, Asara JM, Rabinow L, Perrimon N (2018) The TORC1-regulated CPA complex rewires an RNA processing network to drive autophagy and metabolic reprogramming. Cell Metab 27(1040–1054):e8. https://doi.org/10.1016/j.cmet.2018.02.023
Tang HW, Liao HM, Peng WH, Lin HR, Chen CH, Chen GC (2013) Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev Cell 27:489–503. https://doi.org/10.1016/j.devcel.2013.10.017
Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AI (2014) Nek2 phosphorylates and stabilizes beta-catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell 25:977–991. https://doi.org/10.1091/mbc.E13-06-0349
Bahmanyar S, Kaplan DD, Deluca JG, Giddings TH Jr, O'Toole ET, Winey M, Salmon ED, Casey PJ, Nelson WJ, Barth AI (2008) beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev 22:91–105. https://doi.org/10.1101/gad.1596308
Hung MS, Chen IC, Lung JH, Lin PY, Li YC, Tsai YH (2016) Epidermal growth factor receptor mutation enhances expression of cadherin-5 in lung cancer cells. PLoS ONE 11:e0158395. https://doi.org/10.1371/journal.pone.0158395
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, Huang Y, Yi X, Zhang L (2015) Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol 10:910–923. https://doi.org/10.1097/JTO.0000000000000500
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, C., Peng, S., Li, P. et al. High expression of NEK2 promotes lung cancer progression and drug resistance and is regulated by mutant EGFR. Mol Cell Biochem 475, 15–25 (2020). https://doi.org/10.1007/s11010-020-03854-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03854-z