
Abstract
Activation of epithelial-AR signaling is identified as the major cause of hyperproliferation of the cells during benign and malignant prostate conditions. However, the contribution of stromal-AR is also precarious due to its secretory actions that contribute to the progression of benign and malignant tumors. The present study was aimed to understand the influence of stromal-AR mediated actions on epithelial cells during BPH condition. The secretome (conditioned media-CM) was collected from AR agonist (testosterone-propionate-TP) and antagonist (Nilutamide-Nil) treated BPH patient-derived stromal cells and exposed to BPH epithelial cells. Epithelial cells exhibited increased cell proliferation with the treatment of CM derived from TP-treated stromal cells (TP-CM) but did not support the clonogenic growth of BPH epithelial cells. However, CM derived from Nil-treated stromal cells (Nil-CM) depicted delayed and aggressive BPH epithelial cell proliferation with increased clonogenicity of BPH epithelial cells. Further, decreased AR levels with increased cMyc transcripts and pAkt levels also validated the clonogenic transformation under the paracrine influence of inhibition of stromal-AR. Moreover, the CM of stromal-AR activation imparted positive regulation of basal/progenitor pool through LGR4, β-Catenin, and ΔNP63α expression. Hence, the present study highlighted the restricted disease progression and retains the basal/progenitor state of BPH epithelial cells through the activation of stromal-AR. On the contrary, AR-independent aggressive BPH epithelial cell growth due to paracrine action of loss stromal-AR directs us to reform AR pertaining treatment regimes for better clinical outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Benign prostate hyperplasia (BPH) and prostate cancer (PCa) share similar pathological factors and exhibit hyperproliferation of epithelial cells, alteration of stromal to epithelial ratio with dissimilar cellular histology. BPH does not portray mortality, but it surely deteriorates the quality of life in aging men. Studies have shown that both BPH and PCa coexist in patients and are interlinked for tumor growth [1]. Additionally, a single largest BPH study on Danish men depicted that BPH poses a two- to threefold increased risk to develop PCa [2]. Yet, the coexistence of BPH and PCa in patients has remained paradox due to their molecular differences. One of the key causal factors shared by BPH and PCa is androgen receptor (AR), as during mid-age, atypical activation of AR in epithelial cells becomes the key factor to drive BPH and PCa.
AR plays a critical function in the fetal development of the prostate by influencing AR-positive precursors to fibroblast and smooth muscle cells of urogenital sinus (UGS) and embryonic urogenital mesenchyme (UGM) [3]. Further, fetal androgens acting through stromal-AR maneuvre prostate ductal morphogenesis, epithelial differentiation, and proliferation/apoptosis. Hence, the normal epithelial differentiation and function depend on androgen-mediated stromal paracrine signals [4]. During adulthood, epithelial-AR dominates stromal-AR expression to regulate cellular homeostasis and secretory functions of the prostate gland [5]. Overexpression of epithelial-AR has been reported during BPH and PCa. Though at the cellular level, different expression levels of AR have been observed depending on the stage and grade of the PCa tumors [6]. Nevertheless, stromal-AR also plays an important role alongside with epithelial-AR for disease progression.
In PCa, not only cancer cells but surrounding stroma, known as carcinoma-associated fibroblasts (CAFs), contributes to the survival and proliferation of tumor cells through its secretory anti-apoptotic factors, growth factors, and cytokines [5, 7, 8]. Similarly in BPH also, stromal cells continuously produce cytokines and growth factors to support tumor growth [9]. Thus, growth promotion and inhibition of malignant and non-malignant tumor cells depend on the stromal cell secretory factors. These secretory factors are strongly influenced by activation of AR that regulates the expression and secretion of several cytokines (CCLs, CXCLs, ILs, etc.) and growth factors (IGFs, FGFs, TGFs, etc.) that have paracrine action on epithelial cells during BPH and PCa conditions [10, 11]. Also, it has been observed that gain in stromal-AR in CAFs rescued rapid growth and progression of the cancerous tumors, whereas loss of stromal-AR was directly correlated with advanced pathological stage of PCa [5]. However, the underlying role of stromal-AR influencing the hyperplastic growth of epithelial cells during BPH condition is still obscure.
AR is the central regulatory protein in prostate development and disease pathogenesis. It profoundly functions in epithelial cells during benign and malignant tumors. In addition to epithelia, stromal cell-derived factors are also implicated to regulate prostatic disease progression. Thus, the present study was aimed to explore the role of stromal-AR mediated regulation of the epithelial cell growth and disease progression during BPH condition using patient-derived stromal and epithelial cells. As AR targeted therapies are widely used both for BPH and PCa patients, we also aimed to evaluate the implication of such drugs on stromal epithelial crosstalk in disease progression which will enable us to throw more light on the clinical relevance of AR targeted therapies.
Materials and methods
Cell culture
The protocol for collection and isolation of cells from human BPH patient tissue was approved by the Institutional ethical committee for human research (protocol number: IECHR/2016-07) of the Department of Biochemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara and informed patient consent was acquired to use the surgically excised tissue for the research purposes only. Briefly, after confirmation of histological BPH in surgically excised prostate tissue, tissue chips were subjected to smooth muscle cells (SMCs) isolation. BPH patient-derived SMCs were isolated using the previously described protocol with the modification to purify the stromal cells population from the BPH tissue sample [12]. Isolated SMCs were maintained in DMEM-F12 media (Gibco#12,500-062) supplemented with 1X penicillin–streptomycin (Gibco#15,140-122), 1X Glutamax (Gibco#35,050,061) and 10% fetal bovine serum (FBS; Gibco#10,270-106). Experiments performed with SMCs are on or before passage#6. Previously isolated BPH epithelial cell-line was also maintained in a similar fashion and experimented before passage#20 in charcoal-stripped serum (CSS) [12]. The CSS was prepared using a previously described protocol [13]. Briefly, a single lot of FBS (Gibco#10,270–106) was supplemented with 2% w/v activated charcoal (SRL#55,192) and incubated for 3 h at 4 °C with rotation followed by CSS retrieval through centrifugation and 0.22-μm filter sterilization.
Immunocytochemistry
BPH patient-derived SMCs were grown on a glass coverslip in a 3 cm2 culture plate containing growth medium. The culture media was aspirated, washed with phosphate buffer saline (PBS) and fixed in 2% p-formaldehyde at room temperature (RT) for 10 min followed by permeabilization with 0.1% Triton-X-100 for 5 min. Cells were blocked with the buffer containing 1% bovine serum albumin (BSA) in PBS for 1 h at RT. FITC labeled anti-α smooth muscle actin and PE-labeled anti-vimentin antibodies were prepared in 0.1% BSA containing PBS and treated to SMCs overnight at 4 °C in dark. Primary Antibodies were removed by washing with 0.1% BSA containing PBS. Cells were then mounted to a clean glass slide in mounting medium containing anti-fade (Sigma#S6776) and imaging was performed on a Nikon T200 fluorescence microscope and analyzed the data using NIS-Elements BR software.
Flow cytometry
SMCs were cultured in a 15 cm2 plate till 80% confluency. Cells were scraped and pipetted several times to prepare single-cell suspension in chilled PBS. Around 0.2 × 106 cells were counted and incubated with Anti-CD90 (FITC) and Anti-CD34 (FITC) antibodies separately for 1 h at RT in dark followed by washing. Flow cytometry was performed on FACS Aria III (BD Biosciences, San Jose, CA) instrument and analysis was performed using FlowJo software (BD Biosciences, San Jose, CA).
Conditioned media (CM) preparation from Stromal cell and treatment to BPH epithelial cells
BPH patient-derived cells were serum-starved in DMEM-F12 media for 48 h (hrs). Post serum starvation, SMCs were replenished with serum-free media (SFM) (DMEM/F12 Gibco#12,500-062) containing 10 nM Testosterone Propionate (TP) and 50 μM Nilutamide (Nil) for 48 h to activate and inhibit stromal-AR. Untreated CM (Control-CM), TP-treated CM (TP-CM), and Nil-treated CM (Nil-CM) were collected in a sterile 50 ml tube and centrifuged at 4000 rpm for 30 min at 4 °C to remove cell debris, followed by filtration through 0.22-μm filter. The CM was then stored at − 80 °C until the treatment to epithelial cells and other experimental use.
Cell proliferation assay
Approximately 2500 cells were seeded in a 96-well plate and allowed them to adhere overnight. Treatments of CM or CM containing 10 ng/ml Anti-IGF-1- monoclonal antibody (mAb), 10 nM TP, and 50 µM Nil were given to respective cells. The temporal analysis was performed at 24, 48, and 72 h for each treatment group. For assessing formazan conversion from MTT, the culture media was aspirated from the respective wells and time, followed by supplementation of 0.5 mg/ml MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) (HiMedia#TC191) in SFM. The cells were then incubated for 3 h in a humid incubator containing 5% CO2. MTT media was aspirated post-incubation and formazan crystal in the cells was dissolved in DMSO (Dimethyl sulfoxide) giving a purple colored product. Absorbance was taken at 540 nm using Multiskan™ plate reader (Thermo Scientific). Percent cell proliferation of the cells was calculated with respect to the control group.
Enzyme-linked immunosorbent assay
Collected CM of each treatment group was thawed from − 80 °C and IGF-1 was quantified by sandwich enzyme-linked immunosorbent assay (ELISA) kit (RayBiotech#ELH-IGF-1-1), according to the manufacturer’s instructions.
Gene expression analysis by qPCR
Epithelial cells were serum-starved for 12 h and then treated with CM for 48 h. Cells were then harvested in TRIZOL™ reagent (Invitrogen#15,596,018) for total RNA isolation followed by cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems#4,368,814) as per the manufacturer’s protocol. Gene expression was assessed using SyBr Green (Takara#RR820A) and target gene-specific primers using ABI-7500™ (Applied Biosystems) qPCR system. All real-time qPCR assays were carried out in three independent cDNA syntheses using specific primers as follows:
Sr no | Gene | Sequence (5′–3′) | Product size (BP) | Accession number | |
|---|---|---|---|---|---|
1 | LGR4 | Forward Primer | GAAGAGCTACAATTGGCGGG | 242 | NM_018490.2 |
Reverse Primer | CTGTTGTCATCCAGCCACAG | ||||
2 | β-CATENIN | Forward Primer | GCGCCATTTTAAGCCTCTCG | 183 | NM_001904.3 |
Reverse Primer | AAATACCCTCAGGGGAACAGG | ||||
4 | MYC | Forward Primer | TACAACACCCGAGCAAGGAC | 162 | NM_002467.4 |
Reverse Primer | GAGGCTGCTGGTTTTCCACT | ||||
5 | β-ACTIN | Forward Primer | ACTCTTCCAGCCTTCCTTCC | 101 | NM_001101 |
Reverse Primer | CGTACAGGTCTTTGCGGATG |
Immunoblotting
Cells were scraped in chilled PBS and centrifuged after completion of treatment time. Whole-cell lysates were extracted using laemmli buffer containing and sonicated for total protein extraction. Total protein concentration was determined by the Bradford method and 40 μg of total protein was loaded in each 10% SDS-PAGE gel as described previously [14]. Proteins were transferred to a 0.22-μm nitrocellulose membrane by western blotting. Blocking was performed at RT for 1 h and incubated with specific primary antibody (listed below) overnight at 4 °C. Horseradish peroxidase (HRP) labeled secondary antibody was incubated for 1 h at RT and washed with PBS containing Tween20 (Promega#H5152) and PBS. Blots were developed with Bio-Rad enhanced chemiluminescence reagent on UVTECH™ Cambridge Alliance 4.7 instrument. Captured images were analyzed by measuring band intensities of control and treated groups normalized with loading control using ImageJ Software.
Sr. no | Antibody | Make | Catalog |
|---|---|---|---|
1 | Androgen receptor | Sigma | A9853 |
2 | LGR4 | Thermo scientific | PA527177 |
3 | Total Akt | CST | 4691S |
4 | pAkt(s473) | CST | 4060P |
5 | β-Actin | BD bioscience | 612,657 |
6 | β-Catenin | Sigma | 9AB4500545 |
7 | α-Smooth muscle actin -FITC | Sigma | F3777 |
8 | Vimentin-Cy3 | Sigma | C9080 |
9 | CD90-FITC | BD bioscience | 553,012 |
10 | CD34-FITC | BD bioscience | 560,942 |
11 | ΔNP63 | CST | 67,825 |
Antibody dilutions WB-1:1000; FACS-1:100; ICC-1:40 | |||
Clonogenic assay
Around ~ 20 × 10−3 BPH epithelial cells of (between passage#16–20) for secretome treatment and ~ 2.5 × 10−3 BPH epithelial cells for direct TP/Nil treatment were seeded in a six-well plate with DMEM/F12 media with 10% FBS and incubated overnight at 37 °C for adherence. The media was aspirated and supplemented with respective treatments of stromal secretome and 10 nM TP & 50 μM Nil prepared in 5% charcoal-stripped serum (CSS) for 6 days with regular change of respective treatment media every two days. On the 6th day, treatment media was removed and cells were washed with PBS (Phosphate Buffer Saline). Cells were fixed with 4% formaldehyde for 10 min at RT followed by staining with 0.5% w/v crystal violet for 30 min. The solution was removed and washed with distilled water. The cells were then left to dry at RT. The image of the plate was then taken at 20× image resolution under a phase-contrast microscope.
Statistical analysis
All the experiments are performed thrice independently unless noted separately. All the values in the graph represent Means ± SEM. A comparison between the two groups was performed using t test. The significance between time and treatment variables of the groups was performed with two-way ANOVA and Bonferroni post-test. The statistical significance was considered with the p values ≤ 0.05.
Results
Effect of AR activation on BPH patient-derived stromal cells
AR performs diverse functions in the prostate gland via regulating critical genes in epithelial and stromal cells to perform secretory functions. To understand the crosstalk between stromal and epithelial cells mediated by AR, the stromal cells were isolated and cultured from the surgically excised BPH patient tissue. The isolated cells demonstrated the expression of Vimentin (Red) and α-SMA (Green) confirming the presence of SMCs (Fig. 1a). Further, the flow cytometric evaluation of the expression of 75.5% CD90 and 1.55% CD34 depicted CD90+ve/CD34low population further confirms the presence of SMCs (Fig. 1b). The treatment with AR agonist (10 nM TP) significantly increased the AR protein levels (1.36 ± 0.08 fold; p ≤ 0.05) in SMCs, whereas AR antagonist (50 µM Nil) treatment declined AR protein expression (0.76 ± 0.05 fold; p > 0.05) (Fig. 1c). BPH patient-derived stromal cells showed a significant increase in percent cell proliferation with TP treatment at 24-h (114.65 ± 3.73%; p ≤ 0.01)and 48 h (128.85 ± 3.6%; p ≤ 0.05) and then decreased at 72 h (98.51 ± 6.2; p ≤ 0.05) as compared to control SFM media (100.00 ± 2.2, 109.05 ± 3.3, 120.53 ± 0.3% at 24, 48, 72 h, respectively). However, inhibition of AR with Nil treatment decreased the percent cell proliferation in a time-dependent manner at 24 (82.70 ± 4.13%; p ≤ 0.01), 48 (92.91 ± 2.4%; p ≤ 0.01), 72 (38.63 ± 2.7%; p ≤ 0.05) (Fig. 1d). Thus, activation and inhibition of stromal-AR influences the proliferative potentials of SMCs derived from BPH patient.

Androgens regulates the cell proliferation of BPH patient-derived SMCs. a α-SMA and Vimentin expression were assessed in isolated cells suggesting the presence of SMCs, confirming resolution their identification as stromal cells; image − 20×; Scale bar: 100 μm. b Flow cytometric analysis of CD90 and CD34 in BPH patient-derived SMCs. c AR protein expression in stromal cells upon 10 nM-TP and 50 µM-Nil treatments: Plotted values in bar graph represent Mean ± S.E.M. (n = 3; *p ≤ 0.05). d Assessment of stromal cell proliferation by MTT upon 10 nM-TP and 50 µM- Nil treatments: Bars represent percent cell proliferation of each treatment group at different time intervals. Data represented as Mean ± S.E.M.. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 all groups vs 24-h Control (SFM); @p ≤ 0.05, @@p ≤ 0.01 48-h Control (SFM) vs 48-h treatments; !p ≤ 0.05, !!!!p ≤ 0.0001 72-h Control (SFM) vs 72-h treatments
Activation and inhibition of stromal-AR affect the growth and clonogenicity of BPH epithelial cells
To evaluate the paracrine effect of stromal-derived secretome on epithelial cells, CM was prepared via activation of stromal-AR (TP-CM) and inhibition of stromal-AR (Nil-CM) as described in Fig. 2a. Treatment with TP-CM to BPH epithelial cells demonstrated increased cell proliferation at 48 (122.25 ± 0.54%; p ≤ 0.05) and 72 (151.01 ± 5.94%; p ≤ 0.01) hrs as compared to control-CM treatment (100.00 ± 3.7, 103.91 ± 2.9, 126.81 ± 5.7% at 24, 48 and 72 h, respectively). Whereas, Nil-CM treatment to epithelial cells showed less but significant inhibition of BPH epithelial cell growth at 48 h (91.42 ± 1.9%; p ≤ 0.05) against control-CM and TP-CM treatments. Strikingly, delayed cell proliferation was observed in Nil-CM treatment at 72 h (141.53 ± 2.1%; p = 0.05) (Fig. 2b). On the contrary, direct exposure of 10 nM TP in SFM did not show proliferative effect in BPH epithelial cells. Whereas, direct exposure of 50 µM Nil significantly decreased the BPH epithelial cell growth at 24 (89.5 ± 0.7%; p ≤ 0.05), 48 (66.03 ± 3.6%; p ≤ 0.01), 72 (41.9 ± 2.2%; p ≤ 0.01) hrs as compared to control (100 ± 3.45, 96.1 ± 5.03, 89.4 ± 8.03% at 24, 48 and 72 h, respectively) (Fig. 2c).

Activation and inhibition of stromal-AR alters the cell proliferation and clonogenicity of BPH epithelial cells. a Work plan for secretome preparation and collection, b Cell proliferation of BPH epithelial cells after treatment of Control-CM, TP-CM, and Nil-CM. Data represented as Mean ± S.E.M.; n = 3–4; @p ≤ 0.05 all groups 48-h vs 48-h Control-CM treatment; !p ≤ 0.05 72-h of all groups vs 72-h Control-CM treatment; $p ≤ 0.05 48 h vs 72-h TP-CM treatment; ###p ≤ 0.001 48-h vs 72-h Nil-CM treatment. c Cell proliferation of BPH epithelial cells after treatment of 10 nM TP and 50 µM Nil in SFM. Data represented as Mean ± S.E.M.; n = 3; *p ≤ 0.05 vs 24-h Control; @@p ≤ 0.01 vs 48hrs Control; !!p ≤ 0.01 vs 72hrs Control. d Clonogenic growth of BPH epithelial cells treated with AR agonist and antagonist exposed stromal cells derived CM (Control-CM, TP-CM, and Nil-CM) and images captured at different time intervals; the left brightfield image panel was captured at day 0 (left), day 3 (middle), day 6 (right) and right panel show crystal violet staining on 6th day of respective treatments during clonogenic growth of BPH epithelial cells. e Clonogenic growth of BPH epithelial cells treated with 10 nM TP and 50 µM Nil and images captured at different time intervals; the left panel represents brightfield images captured at day 0 (left), day 3 (middle), day 6 (right) and right panel shows crystal violet staining on 6th day of respective treatments during clonogenic growth of BPH epithelial cells. n = 2, image − 4× and 20×; Scale bar: 100 μm
Further, we investigated the potentials of the secretome to induce clonogenicity in epithelial cells. Results from clonogenic assay showed cell cluster formation with CM treatments of all the groups until day 3. Strikingly, complete epithelial cell death was observed in the case of TP-CM treatment as compared to Control-CM and Nil-CM treatments on the 6th day of clonogenic assay. In contrast, Nil-CM treatment exhibited higher clonogenic expansion of BPH epithelial cells on the 6th day (Fig. 2d). Moreover, direct treatment of 10 nM TP to BPH epithelial cells formed large epithelial cell clusters, whereas 50 μM Nil exposed cell clusters were less in number and similar in size to untreated control. (Fig. 2e) Collectively, inhibition of stromal-AR imparted a high capacity for clonogenic expansion of epithelial cells. In contrast, activation of stromal-AR counters the clonogenic growth of epithelial cells. Thus, the results demonstrated a completely antagonistic effect of stromal secretome derived from the modulation of AR and the direct action of AR in BPH epithelial cells.
Stromal-AR does not affect IGF-1 secretion in BPH patient-derived stromal cells
Expression and secretion of many peptides from stromal cells can regulate epithelial cell growth under the control of AR. One of the key growth factors secreted by prostate stroma is IGF-1 that has a strong mitogenic effect on epithelial cells. The quantitative estimation of IGF-1 levels depicted no significant change between Control-CM (2.58 ± 0.15 pg), TP-CM (3.26 ± 0.38 pg; p > 0.05), and Nil-CM (2.58 ± 0.21 pg; p > 0.05) groups (Fig. 3a). Further, treatment of Control-CM, TP-CM, and Nil-CM supplemented with Anti-IGF-1-mAb, depicted a significant decline in percent cell proliferation of epithelial cells with increasing incubation time. (Control-CM against Control-CM + Anti-IGF-1-mAb (24-h: 100.00 ± 2.13 and 89.82 ± 3.33%; 48-h: 105.17 ± 5.23 and 75.07 ± 3.7%; 72-h: 132.46 ± 1.2 and 77.12 ± 8.8%), TP-CM against TP-CM + Anti-IGF-1-mAb (24-h: 107.26 ± 3.0 and 104.94 ± 9.3%; 48-h: 116.17 ± 6.1 and 85.12 ± 1.5%; 72-h: 151.00 ± 5.9 and 86.19 ± 3.2%) and Nil-CM against Nil-CM + Anti-IGF-1-mAb (24-h: 98.39 ± 1.8 and 175.2 ± 6.0%; 48-h: 91.4 ± 1.9and 79.9 ± 0.7%; 72-h:141.53 ± 2.1 and 79.54 ± 1.7%) (Fig. 3b). Thus, the secretory levels of IGF-1 were not affected by the alterations in stromal-AR activity in BPH patient-derived stromal cells. However, results suggest a crucial role of stromal secreted IGF-1 on epithelial cell proliferation.

Stromal-AR does not affect the secretion of IGF-1 in BPH patient-derived SMCs. a Quantitative estimation of IGF-1 in Control-CM, TP-CM, and Nil-CM groups by ELISA; Mean ± S.E.M.; n = 3; b Cell proliferation of BPH epithelial cells after treatment of 10 ng/ml anti-IGF-1-mAB in each secretome Control-CM, TP-CM, and Nil-CM and compared with their respective treatment. Data represents Mean ± S.E.M.; n = 3;***p ≤ 0.001 all groups compared with SFM Control; $p ≤ 0.05, $$$p ≤ 0.001 Control-CM vs Control-CM + IGF-1-mAb, ##p ≤ 0.01, ###p ≤ 0.001 TP-CM vs TP-CM + IGF-1-mAb, !p ≤ 0.05, !!p ≤ 0.01, !!!p ≤ 0.001 Nil-CM vs Nil-CM + IGF-1-mAb
Stromal-AR inhibition influences cell survival and clonogenicity via altering AR, Akt, and cMyc expression
To evaluate the levels of epithelial-AR, AR protein expression was assessed. AR protein levels were significantly upregulated with TP-CM treatment (1.114 ± 0.01-fold; p ≤ 0.01) and downregulated with Nil-CM treatment (0.80 ± 0.06-fold; p ≤ 0.05). Apart from AR, IGF-1 plays a substantial role in disease progression via activation of Akt signaling. Evaluation of total Akt and phosphorylated Akt (pAktS473) depicted a profound increase in BPH epithelial cells upon both TP-CM (Total Akt: 3.53 ± 0.6-fold; p ≤ 0.05 and pAkt: 1.24 ± 0.08-fold; p ≤ 0.05, respectively) and Nil-CM (Total Akt: 3.71 ± 0.8; p ≤ 0.05 and pAkt: 1.46 ± 0.06-fold; p ≤ 0.05, respectively) treatments as compared to Control-CM group. However, Nil-CM had significantly higher pAkt (p ≤ 0.05) than the TP-CM group supporting higher cell proliferation and survival capability (Fig. 4a). A previous report suggested increasing cMyc expression was determined as a vital oncogenic event during the early stage of PCa [15]. As we observed increased clonogenicity in BPH epithelial cells with Nil-CM treatment, we assessed the expression of cMyc in BPH epithelial cells. The significant increase in cMyc transcript levels was detected in BPH epithelial cells treated with Nil-CM (5.63 ± 1.4 folds; p ≤ 0.05) against Control-CM treatment with no change in TP-CM group (0.84 ± 0.06-fold; p > 0.05) (Fig. 4b). Collectively, BPH epithelial cells exhibited increased pAkt and cMyc with decreased AR levels due to the paracrine effect of CM derived by stromal-AR inhibition, supporting cell survival, and enhanced clonogenicity in epithelial cells.

Inhibition of stromal-AR affects Myc, AR, and Akt levels in BPH epithelial cells through secreted factors. a Immunoblot images and graphs representing protein expression of AR, tAKt and pAkt normalized with β-Actin in BPH epithelial cells upon Control-CM, TP-CM, and Nil-CM treatments; Data represented as Mean ± S.E.M.; n = 3; *p ≤ 0.05, **p < 0.01 as compared to control-CM; @p ≤ 0.05, TP-CM vs Nil-CM. b Graph representing transcript levels of cMyc in BPH epithelial cells upon Control-CM, TP-CM, and Nil-CM treatments; Data represented as Mean ± S.E.M.; n = 4; *p ≤ 0.05 as compared to control-CM
Secretory actions of stromal-AR activation positively influence the basal/progenitor state of the BPH epithelial cells
Previously isolated BPH epithelial cells used in this study depicted the expression of pluripotency and basal stem cell markers [16]. As secreted factors from prostate stroma regulate the basal/progenitor state of the epithelial cells [17], we intended to explore if the basal/progenitor populations are involved in clonogenic growth in BPH. Hence, LGR4 and ΔNP63α basal/progenitor markers were investigated in BPH epithelial cells with the treatment of CM derived from activation and inhibition of stromal-AR. The transcript levels of LGR4 (12.55 ± 3.1fold; p ≤ 0.05) and its downstream effector, β-Catenin (3.59 ± 0.7-fold; p ≤ 0.05) showed upregulation in BPH epithelial cells with TP-CM treatment as compared to Control-CM and Nil-CM treatments (Fig. 5a). Additionally, the protein expression of LGR4 (2.2 ± 0.2 folds; p ≤ 0.05) and ΔNP63α (1.8 ± 0.16 folds; p ≤ 0.05) were significantly increased with TP-CM treatment no significant alterations in LGR4 (0.74 ± 0.04-fold; p > 0.05) and ΔNP63α (0.98 ± 0.25-fold; p > 0.05) in Nil-CM treated group in BPH epithelial cells (Fig. 5b). Thus, the results suggest that activation of stromal-AR derived CM supports the increased levels of LGR4/β-Catenin/ΔNP63α suggesting a positive effect on basal/progenitor markers.

Activation of stromal-AR influences the basal/progenitor state of BPH epithelial cells through secreted factors. a Graphs representing transcript levels of LGR4 and β-Catenin in BPH epithelial cells upon Control-CM, TP-CM, and Nil-CM treatments; Data represented as Mean ± S.E.M.; n = 4; *p ≤ 0.05,**p ≤ 0.01 as compared to control- CM. b Immunoblot images and graphs representing protein expression of AR, LGR4 and ΔNP63α normalized with β-Actin in BPH epithelial cells upon Control-CM, TP-CM and Nil-CM treatments; Data represented as Mean ± S.E.M.; n = 3; *p ≤ 0.05, as compared to control-CM; @p ≤ 0.05, TP-CM vs Nil-CM
Discussion
Crosstalk between stromal and epithelial cells in the prostate tumor microenvironment has immense implications in tumor growth. The involvement of stroma has been identified as a key regulator of PCa tumors [18]. The cells of cancerous stroma, known as cancer-associated fibroblasts (CAFs), can aggressively induce PCa progression, metastasis, and therapy resistance in patients [19, 20]. However, the role of AR in stromal-epithelial crosstalk is not largely explored during BPH condition. Hence, to explore the role of stromal-AR on epithelial cells, we have isolated fibroblasts from BPH patients expressing Vimentin and α-SMA, ensuring the presence of SMCs, the major cell type of adult prostate stroma [21, 22]. The majority of these isolated SMCs are CD90+ve/CD34−ve and interestingly, CD90+ve stromal cells are in direct contact with the epithelial cells surrounding the acini, that express growth factors and genes related to cell motility, developmental process, and androgen biosynthesis to promote epithelial growth [23]. Hence, the isolated CD90+ve SMCs in the present study must be directly influencing the epithelial cells in a paracrine manner.
AR is the master driver of PCa through epithelial cell proliferation contributing to tumor growth [24]. In agreement with Leimgruber’s report, stimulation of TP in the primary cultures of SMCs enhanced their cell proliferation [25]. Additionally, stimulation of androgen in primary fibroblasts of healthy human subjects showed increased AR protein expression [26]. Previously, tissue recombination and mouse knockout depicted that activation of stromal-AR promotes epithelial growth during gland development [27, 28]. Further, activation and inhibition of stromal-AR have substantial involvement in the disease progression of the prostate gland [5]. These evidences support that stromal cell secretome derived through stromal-AR activation is vitally involved in fetal development of the gland PCa progression also. The coculture between AR-negative stromal cells and PC3 cells resulted in enhancement of the growth rate of PC-3 cells compared to individually grown PC-3 cells [29]. Similarly, the coculture of AR expressing CAFs with PCa cell-lines showed decreased invasive potentials of the cancer cells [30,31,32]. Moreover, a greater decrease of stromal-AR has been reported in the cells of the cancerous tissue as compared to BPH tissue which was also associated with increasing tumor grade [29]. Further, androgen deprivation and loss of stromal-AR alters paracrine actions by promoting the invasiveness via altering the extracellular matrix in PCa patients [33, 34]. These evidences support the present data where inhibition of stromal-AR derived from BPH patient imposed clonogenic growth in the BPH epithelial cells through patient-derived stromal cell secretory factors. Moreover, this is the first report to highlight two completely diverse effects of cell-specific AR expression in BPH condition; where epithelial-AR activation increases clonogenicity, but stromal-AR activation decreases clonogenicity in the BPH patients.
One of the key mitotic growth factors synthesized and secreted by stroma is IGF-1, and its receptor (IGF-1R) expressed on prostate epithelial cells that activate Akt signaling [35]. The bioavailability of serum IGF-1 was found to be increased in BPH patients and its overexpression caused the neoplastic transformation of the murine prostate epithelium [36, 37]. In the present study, IGF-1 secretion from BPH stromal cells derived CM was not affected due to alterations in stromal-AR. Yet, trapping secreted IGF-1 with mAb significantly reduced the growth of BPH epithelial cells suggesting a significant contribution of stromal secreted IGF-1in cell proliferation. Since BPH stroma does not have as extensive chromatin changes as in CAFs, AR may not be able to exert similar regulatory action. Additionally, AR was also found to regulate other secreted factors as well [38], which may involve in the survival and clonogenicity of BPH epithelial cells.
We showed that the secretome of inhibited stromal-AR induces cell survival and clonogenicity which is corroborated with increased pAkt and cMyc and decreased AR levels. Previously, higher levels of cMyc expression were detected in PCa patients as compared to BPH patients [39, 40] and bestow an androgen-independent and invasive PCa tumor growth [41, 42]. Previously, Williams et al. showed that increased cMyc expression in BPH epithelium is sufficient to induce carcinogenesis [43]. The initiation, advancement, and reoccurrence of PCa are often found to be driven by cMyc proto-oncogene [44, 45]. Further, Myc overexpression also reduced AR signaling in mouse xenografts and drives resistance against AR inhibitors and profoundly occupied the chromatin sites of decreasing AR levels and promotes AR-independent tumor growth [46,47,48]. The bigenic (mpAkt/Hi-Myc) mice model discovered that Myc promoted Akt to form drug-resistant PCa and increased pAkt levels inhibits apoptosis and supports the survival and growth of tumor cells in PCa [49, 50]. The majority of the stromal secreted growth factors and cytokines acts via activation of the Akt pathway for cell survival and proliferation [51, 52]. Since AR, cMyc, and Akt expressions are interlinked in PCa survival and tumorigenesis, their role is explicitly relevant in this study also. This study, therefore, corroborates cell survival and clonogenic growth of BPH epithelial cells, which was mediated through paracrine action of stromal-AR inhibition, which induced the upregulation of oncogene cMyc and Akt. Thus, the progressive loss of stromal-AR in BPH patients enhances the risk of oncogenic activation and cell proliferation/survival pathways, which can aid malignant changes in the tissue.
The basal/progenitor cells profoundly contribute to the development of BPH and PCa [16]. AR expression is found negative or low in basal/progenitor cells, and increasing AR expression leads to differentiation of basal/progenitor cells [53]. Furthermore, stromal secretory factors can regulate resident basal/progenitor cell populations in the prostate gland [54]. Kwon et al. recently discovered that coculturing stromal cells with basal stem cells increase basal phenotype and organoid forming activity of these cells through its paracrine activity [23]. Stromal cells produce multiple Wnt ligands that induce Wnt/β-Catenin signaling in prostate basal stem cells [55] which are regulated by a key stemness marker, LGR4 [56, 57]. LGR4 expression was depicted in both human basal and luminal cells and its overexpression drives AR signaling activation with increased cell survival during prostate tumorigenesis in PCa cell-lines [58, 59]. In this study, we have depicted that LGR4 and its downstream effectors AR and β-Catenin were increased in epithelial cells upon AR activated stromal-AR secretome treatment. Increased β-Catenin can bind to the promoter of ΔNp63α and upregulate its expression [60, 61]. And ΔNp63α is the predominant isotype of P63 that is specifically expressed in prostate basal stem cells to regulate the pool of the basal/progenitor population [62]. Thus, the upregulation of ΔNp63α in BPH epithelial cells supports positive regulation through the activation of stromal-AR secretome. The present study exhibited the first key evidence on stromal-AR mediated positive regulation of ΔNp63α and LGR4/β-Catenin in basal/progenitor cells of BPH condition, which also rationalizes the pathological condition where increased basal/progenitor cells have been found due to expression of stromal-AR in the BPH tissue.
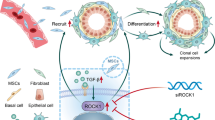
The current study reveals the regulatory role of AR that modulates the fate of epithelial cells via adjacently residing stromal cells in BPH condition. The study highlights the first evidence on activation of stromal-AR that maintains the basal state of the epithelial cells via LGR4, β-Catenin, and ΔNP63α expression but also limits cell proliferation and inhibits clonogenic expansion in epithelial cells during BPH condition. On the contrary, secretome derived from stromal-AR inhibition, cause cell survival, and enhanced clonogenicity through cMyc and Akt leading to the hyperproliferative changes in BPH epithelial cells (Fig. 6). Although IGF-1 contributed to epithelial proliferation, the role of other secretory factors remained elusive. It has been reported that loss of AR is substantially more in PCa than in BPH patients, which is associated with poor outcome and relapse in PCa patients [32, 63]. Moreover, instances where BPH often coexist pathologically in a PCa patient [2, 64], stromal-AR has found to be lost in the tissue surrounding the PCa tumor, yet partially expressed surrounding the BPH tissue [64]. Hence, this study also validates the failure of androgen deprivation therapies in patients with coexisting BPH and PCa where loss or inhibition of stromal-AR in the BPH stroma can augment the growth or relapse of the PCa tumor. Hence, this in vitro study provides an insight into the complex role of stromal-AR loss that accomplice the tumorous growth of BPH epithelial cells that could acquire malignant changes with androgen deprivation therapies.

Stromal-AR exhibit diverse effects on BPH epithelial cells through secreted factors. Activation of stromal-AR limits the growth of the epithelial cells and regulates basal/progenitors. In contrast, inhibition of stromal-AR promotes AR-independent growth of the BPH epithelial cells with increased pAkt and cMyc expression, making them prone to malignant transformation
Conclusion
Our study provides substantial evidence that stromal-AR activation protects the epithelial cells to acquire proliferative changes during BPH condition. Also, loss of stromal-AR and/or use of anti-androgens may bring an undesirable risk of aggressive tumor growth with malignant transformation of BPH epithelial cells. Thus, our study suggests the need for identifying the intriguing roles of stromal-AR and carefully designing treatment regimes of AR for better clinical outcomes in BPH and/or PCa patients.
References
Alcaraz A, Hammerer P, Tubaro A, Schroder FH, Castro R (2009) Is there evidence of a relationship between benign prostatic hyperplasia and prostate cancer? Findings of a literature review. Eur Urol 55(4):864–873. https://doi.org/10.1016/j.eururo.2008.11.011
Orsted DD, Bojesen SE, Nielsen SF, Nordestgaard BG (2011) Association of clinical benign prostate hyperplasia with prostate cancer incidence and mortality revisited: a nationwide cohort study of 3,009,258 men. Eur Urol 60(4):691–698. https://doi.org/10.1016/j.eururo.2011.06.016
Cunha GR, Chung LW, Shannon JM, Reese BA (1980) Stromal-epithelial interactions in sex differentiation. Biol Reprod 22(1):19–42
Simian M, Hirai Y, Navre M, Werb Z, Lochter A, Bissell MJ (2001) The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development 128(16):3117–3131
Singh M, Jha R, Melamed J, Shapiro E, Hayward SW, Lee P (2014) Stromal androgen receptor in prostate development and cancer. Am J Pathol 184(10):2598–2607
Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, Liu B, Tang GW, Zhang D, Tracz A, Jeter C, Rycaj K, Calhoun-Davis T, Huang J, Rubin MA, Beltran H, Shen J, Chatta G, Puzanov I, Mohler JL, Wang J, Zhao R, Kirk J, Chen X, Tang DG (2018) Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun 9(1):3600. https://doi.org/10.1038/s41467-018-06067-7
Shiao SL, Chu GC, Chung LW (2016) Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett 380(1):340–348. https://doi.org/10.1016/j.canlet.2015.12.022
Gascard P, Tlsty TD (2016) Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev 30(9):1002–1019. https://doi.org/10.1101/gad.279737.116
Schauer IG, Rowley DR (2011) The functional role of reactive stroma in benign prostatic hyperplasia. Differ Res Biol Divers 82(4–5):200–210. https://doi.org/10.1016/j.diff.2011.05.007
Zhu M-L, Kyprianou N (2008) Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer 15(4):841–849
Izumi K, Mizokami A, Lin WJ, Lai KP, Chang C (2013) Androgen receptor roles in the development of benign prostate hyperplasia. Am J Pathol 182(6):1942–1949. https://doi.org/10.1016/j.ajpath.2013.02.028
Prajapati A, Gupta S, Bhonde R, Gupta S (2014) Pluripotent stem cell within the prostate could be responsible for benign prostate hyperplasia in human. J Stem Cell Res Ther 4(164):2
Gulick T (1997) Transfection using DEAE-dextran. Curr Protoc Mol Biol 40(1):9–2
Chruvattil R, Banerjee S, Nath S, Machhi J, Kharkwal G, Yadav MR, Gupta S (2017) Dexamethasone alters the appetite regulation via induction of hypothalamic insulin resistance in rat brain. Mol Neurobiol 54(9):7483–7496. https://doi.org/10.1007/s12035-016-0251-2
Imura M, Kojima Y, Kubota Y, Hamakawa T, Yasui T, Sasaki S, Hayashi Y, Kohri K (2012) Regulation of cell proliferation through a KIT-mediated mechanism in benign prostatic hyperplasia. Prostate 72(14):1506–1513. https://doi.org/10.1002/pros.22500
Prajapati A, Gupta S, Mistry B, Gupta S (2013) Prostate stem cells in the development of benign prostate hyperplasia and prostate cancer: emerging role and concepts. Biomed Res Int 2013:107954. https://doi.org/10.1155/2013/107954
Li Q, Alsaidan OA, Rai S, Wu M, Shen H, Beharry Z, Almada LL, Fernandez-Zapico ME, Wang L, Cai H (2018) Stromal Gli signaling regulates the activity and differentiation of prostate stem and progenitor cells. J Biol Chem 293(27):10547–10560. https://doi.org/10.1074/jbc.RA118.003255
Schroeder FH, Westerhof M, Bosch RJ, Kurth KH (1986) Benign prostatic hyperplasia treated by castration or the LH-RH analogue buserelin: a report on 6 cases. Eur Urol 12(5):318–321. https://doi.org/10.1159/000472646
Kyprianou N, Davies P (1986) Association states of androgen receptors in nuclei of human benign hypertrophic prostate. Prostate 8(4):363–380. https://doi.org/10.1002/pros.2990080408
Feneley MR, Puddefoot JR, Xia S, Sowter C, Slavin G, Kirby RS, Vinson GP (1995) Zonal biochemical and morphological characteristics in BPH. Br J Urol 75(5):608–613. https://doi.org/10.1111/j.1464-410x.1995.tb07418.x
Gabbiani G, Schmid E, Winter S, Chaponnier C, De Ckhastonay C, Vandekerckhove J, Weber K, Franke WW (1981) Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci USA 78(1):298–302
Gravina GL, Mancini A, Ranieri G, Di Pasquale B, Marampon F, Di Clemente L, Ricevuto E, Festuccia C (2013) Phenotypic characterization of human prostatic stromal cells in primary cultures derived from human tissue samples. Int J Oncol 42(6):2116–2122. https://doi.org/10.3892/ijo.2013.1892
Kwon OJ, Zhang Y, Li Y, Wei X, Zhang L, Chen R, Creighton CJ, Xin L (2019) Functional heterogeneity of mouse prostate stromal cells revealed by single-cell RNA-seq. iScience 13:328–338. https://doi.org/10.1016/j.isci.2019.02.032
Zhou Y, Bolton EC, Jones JO (2015) Androgens and androgen receptor signaling in prostate tumorigenesis. J Mol Endocrinol 54(1):R15–29. https://doi.org/10.1530/JME-14-0203
Leimgruber C, Quintar AA, Garcia LN, Petiti JP, De Paul AL, Maldonado CA (2013) Testosterone abrogates TLR4 activation in prostate smooth muscle cells contributing to the preservation of a differentiated phenotype. J Cell Physiol 228(7):1551–1560. https://doi.org/10.1002/jcp.24314
Cano P, Godoy A, Escamilla R, Dhir R, Onate SA (2007) Stromal-epithelial cell interactions and androgen receptor-coregulator recruitment is altered in the tissue microenvironment of prostate cancer. Cancer Res 67(2):511–519. https://doi.org/10.1158/0008-5472.CAN-06-1478
Yu S, Zhang C, Lin CC, Niu Y, Lai KP, Chang HC, Yeh SD, Chang C, Yeh S (2011) Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 71(5):517–524. https://doi.org/10.1002/pros.21264
Yu S, Yeh CR, Niu Y, Chang HC, Tsai YC, Moses HL, Shyr CR, Chang C, Yeh S (2012) Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 72(4):437–449. https://doi.org/10.1002/pros.21445
Li Y, Li CX, Ye H, Chen F, Melamed J, Peng Y, Liu J, Wang Z, Tsou HC, Wei J, Walden P, Garabedian MJ, Lee P (2008) Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J Cell Mol Med 12(6B):2790–2798. https://doi.org/10.1111/j.1582-4934.2008.00279.x
Cioni B, Nevedomskaya E, Melis MHM, van Burgsteden J, Stelloo S, Hodel E, Spinozzi D, de Jong J, van der Poel H, de Boer JP, Wessels LFA, Zwart W, Bergman AM (2018) Loss of androgen receptor signaling in prostate cancer-associated fibroblasts (CAFs) promotes CCL2- and CXCL8-mediated cancer cell migration. Mol Oncol 12(8):1308–1323. https://doi.org/10.1002/1878-0261.12327
Leach DA, Buchanan G (2017) Stromal androgen receptor in prostate cancer development and progression. Cancers 9(1):10. https://doi.org/10.3390/cancers9010010
Leach DA, Need EF, Toivanen R, Trotta AP, Palethorpe HM, Tamblyn DJ, Kopsaftis T, England GM, Smith E, Drew PA, Pinnock CB, Lee P, Holst J, Risbridger GP, Chopra S, DeFranco DB, Taylor RA, Buchanan G (2015) Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 6(18):16135–16150. https://doi.org/10.18632/oncotarget.3873
Wikstrom P, Marusic J, Stattin P, Bergh A (2009) Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 69(8):799–809. https://doi.org/10.1002/pros.20927
Kuniyasu H, Chihara Y, Kondo H, Ohmori H, Ukai R (2003) Amphoterin induction in prostatic stromal cells by androgen deprivation is associated with metastatic prostate cancer. Oncol Rep 10(6):1863–1868
Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L (2015) Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis 2(1):13–25
Niu Y-N, Xia S-J (2009) Stroma–epithelium crosstalk in prostate cancer. Asian J Androl 11(1):28
Saikali Z, Setya H, Singh G, Persad S (2008) Role of IGF-1/IGF-1R in regulation of invasion in DU145 prostate cancer cells. Cancer Cell Int 8(1):10
Yamamoto H, Masters JR, Dasgupta P, Chandra A, Popert R, Freeman A, Ahmed A (2012) CD49f is an efficient marker of monolayer- and spheroid colony-forming cells of the benign and malignant human prostate. PLoS ONE 7(10):e46979. https://doi.org/10.1371/journal.pone.0046979
Le Magnen C, Bubendorf L, Ruiz C, Zlobec I, Bachmann A, Heberer M, Spagnoli GC, Wyler S, Mengus C (2013) Klf4 transcription factor is expressed in the cytoplasm of prostate cancer cells. Eur J Cancer 49(4):955–963. https://doi.org/10.1016/j.ejca.2012.09.023
Bhowal A, Majumder S, Ghosh S, Basu S, Sen D, Roychowdhury S, Sengupta S, Chatterji U (2017) Pathway-based expression profiling of benign prostatic hyperplasia and prostate cancer delineates an immunophilin molecule associated with cancer progression. Sci Rep 7(1):9763. https://doi.org/10.1038/s41598-017-10068-9
Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH (2003) Myc confers androgen-independent prostate cancer cell growth. J Clin Invest 112(11):1724–1731. https://doi.org/10.1172/JCI19035
Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL (2003) Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4(3):223–238
Williams K, Fernandez S, Stien X, Ishii K, Love HD, Lau YF, Roberts RL, Hayward SW (2005) Unopposed c-MYC expression in benign prostatic epithelium causes a cancer phenotype. Prostate 63(4):369–384. https://doi.org/10.1002/pros.20200
Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, McLeod DG, Srivastava S, Petrovics G (2010) Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis 13(4):311–315. https://doi.org/10.1038/pcan.2010.31
Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM (2010) MYC and prostate cancer. Genes Cancer 1(6):617–628. https://doi.org/10.1177/1947601910379132
Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS (2016) N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30(4):563–577. https://doi.org/10.1016/j.ccell.2016.09.005
Barfeld SJ, Urbanucci A, Itkonen HM, Fazli L, Hicks JL, Thiede B, Rennie PS, Yegnasubramanian S, DeMarzo AM, Mills IG (2017) c-Myc antagonises the transcriptional activity of the androgen receptor in prostate cancer affecting key gene networks. EBioMedicine 18:83–93. https://doi.org/10.1016/j.ebiom.2017.04.006
Hoang DT, Iczkowski KA, Kilari D, See W, Nevalainen MT (2017) Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: opportunities for therapeutic targeting from multiple angles. Oncotarget 8(2):3724–3745. https://doi.org/10.18632/oncotarget.12554
Clegg NJ, Couto SS, Wongvipat J, Hieronymus H, Carver BS, Taylor BS, Ellwood-Yen K, Gerald WL, Sander C, Sawyers CL (2011) MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLoS ONE 6(3):e17449. https://doi.org/10.1371/journal.pone.0017449
Das TP, Suman S, Alatassi H, Ankem MK, Damodaran C (2016) Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis 7:e2111. https://doi.org/10.1038/cddis.2015.403
Siu MK, Suau F, Chen WY, Tsai YC, Tsai HY, Yeh HL, Liu YN (2016) KLF4 functions as an activator of the androgen receptor through reciprocal feedback. Oncogenesis 5(12):e282. https://doi.org/10.1038/oncsis.2016.79
Lin VK, Wang SY, Vazquez DV, Xu C, Zhang S, Tang L (2007) Prostatic stromal cells derived from benign prostatic hyperplasia specimens possess stem cell like property. Prostate 67(12):1265–1276. https://doi.org/10.1002/pros.20599
Vander Griend DJ, Litvinov IV, Isaacs JT (2014) Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci 10(6):627–642. https://doi.org/10.7150/ijbs.8756
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P (2010) Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 70(17):6945–6956
Wei X, Zhang L, Zhou Z, Kwon OJ, Zhang Y, Nguyen H, Dumpit R, True L, Nelson P, Dong B, Xue W, Birchmeier W, Taketo MM, Xu F, Creighton CJ, Ittmann MM, Xin L (2019) Spatially restricted stromal wnt signaling restrains prostate epithelial progenitor growth through direct and indirect mechanisms. Cell Stem Cell 24(5):753–768. https://doi.org/10.1016/j.stem.2019.03.010
Luo W, Rodriguez M, Valdez JM, Zhu X, Tan K, Li D, Siwko S, Xin L, Liu M (2013) Lgr4 is a key regulator of prostate development and prostate stem cell differentiation. Stem Cells 31(11):2492–2505. https://doi.org/10.1002/stem.1484
Qian Y, Liu S, Guan Y, Pan H, Guan X, Qiu Z, Li L, Gao N, Zhao Y, Li X, Lu Y, Liu M, Li D (2013) Lgr4-mediated Wnt/beta-catenin signaling in peritubular myoid cells is essential for spermatogenesis. Development 140(8):1751–1761. https://doi.org/10.1242/dev.093641
Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RGJ, Cuppen E, Chen Y, Sawyers CL, Clevers HC (2014) Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159(1):163–175. https://doi.org/10.1016/j.cell.2014.08.017
Zhang J, Li Q, Zhang S, Xu Q, Wang T (2016) Lgr4 promotes prostate tumorigenesis through the Jmjd2a/AR signaling pathway. Exp Cell Res 349(1):77–84. https://doi.org/10.1016/j.yexcr.2016.09.023
Chu WK, Dai PM, Li HL, Chen JK (2008) Glycogen synthase kinase-3beta regulates DeltaNp63 gene transcription through the beta-catenin signaling pathway. J Cell Biochem 105(2):447–453. https://doi.org/10.1002/jcb.21839
Ruptier C, De Gasperis A, Ansieau S, Granjon A, Taniere P, Lafosse I, Shi H, Petitjean A, Taranchon-Clermont E, Tribollet V, Voeltzel T, Scoazec JY, Maguer-Satta V, Puisieux A, Hainaut P, Cavard C, Caron de Fromentel C (2011) TP63 P2 promoter functional analysis identifies beta-catenin as a key regulator of DeltaNp63 expression. Oncogene 30(46):4656–4665. https://doi.org/10.1038/onc.2011.171
Di Como CJ, Urist MJ, Babayan I, Drobnjak M, Hedvat CV, Teruya-Feldstein J, Pohar K, Hoos A, Cordon-Cardo C (2002) p63 expression profiles in human normal and tumor tissues. Clin Cancer Res 8(2):494–501
Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, Delprado W, Stricker PD, Grygiel JJ, Sutherland RL (2001) Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res 61(2):423–427
Olapade-Olaopa EO, MacKay EH, Taub NA, Sandhu DP, Terry TR, Habib FK (1999) Malignant transformation of human prostatic epithelium is associated with the loss of androgen receptor immunoreactivity in the surrounding stroma. Clin Cancer Res 5(3):569–576
Acknowledgements
We thank Dr. Ajit Gohil, Dept. of General Surgery, GMERS Hospital, Gotri, Vadodara for providing BPH patient tissue samples for the study. We also thank Dr. Vikram Sarabhai Institute of Cell and Molecular Biology for the central instrumentation facility. GC received financial support from University Grants Commission (UGC), Govt. of India, New Delhi for pursuing this work.
Author information
Authors and Affiliations
Contributions
GC and SG conceptualized the project. GC performed & analyzed the experiments. AM contributed to perform experiments and data analysis; GC wrote the manuscript. SG supervised the project and revised the manuscript for submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Informed consent
Informed consent was obtained from the BPH patients included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chauhan, G., Mehta, A. & Gupta, S. Stromal-AR influences the growth of epithelial cells in the development of benign prostate hyperplasia. Mol Cell Biochem 471, 129–142 (2020). https://doi.org/10.1007/s11010-020-03773-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03773-z