
Abstract
This study was aimed to explore the molecular mechanism of macrophage polarization and its effect on glioma progression. THP1 cells were cocultured in conditioned medium from U87 human glioblastoma cells to simulate the glioma microenvironment. The expression of miR-32 and PTEN in THP1 cells was detected by real-time PCR. A luciferase reporter assay was conducted to confirm the target relation between miR-32 and PTEN. Western blot assays and ELISA were performed to detect PTEN, M2 macrophage-specific markers, PI3K/AKT signaling proteins, and apoptosis-related proteins. U87 cell proliferation was evaluated by CCK-8 and colony forming assays, and the migration ability of the cells was evaluated by Transwell and wound healing assays. The U87 culture supernatant promoted the M2 phenotype of THP1 cells. miR-32 was upregulated and PTEN was downregulated in THP1 cells with the M2 phenotype in the glioma microenvironment. Luciferase assays confirmed that PTEN expression was suppressed by miR-32 through interaction with the 3′UTR of PTEN. Overexpression of miR-32 suppressed PTEN expression in THP1 cells. Overexpression of miR-32 or downregulation of PTEN promoted the expression of M2 macrophage-specific markers, thereby enhancing M2 macrophage polarization. Additionally, miR-32 inhibited THP1 cell apoptosis via suppressing the PI3K/AKT signaling pathway. Most importantly, the proliferation and migration capacities of U87 cells treated with the THP1 culture supernatant after miR-32 overexpression were enhanced, and these effects could be reversed by cotransfection with pcDNA3.1-PTEN. miR-32 negatively modulates PTEN, thereby promoting M2 macrophage transformation through PI3K/AKT signaling, enhancing glioma proliferation and migration abilities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas are among the most common type of primary brain tumors in adults and account for over 70% of malignant brain tumors, with an incidence rate of 3.2 per 100,000 population [1]. Gliomas arise from glial or precursor cells of neuroectodermal origin, including astrocytomas, oligodendrogliomas, and ependymomas [2]. Gliomas are classified into circumscribed gliomas (WHO grade I) and diffusely infiltrating gliomas (WHO grades II–IV; whether astrocytic or oligodendroglial) according to the 2016 version of the WHO guidelines [3]. In spite of the advances in surgery, radiotherapy and chemotherapy for gliomas over the last 30 years, no significant increase in patient survival has been achieved for glioma patients [4]. Therefore, the mechanisms that underlie the occurrence and development of gliomas require elucidation to aid in the development of therapies. Molecular targeted therapies are currently being studied extensively in many cancers, and microRNAs are considered hopeful candidates for using in diagnostic and prognostic methods.
The tumorigenesis, progression and invasion of a tumor depend on the surrounding tumor microenvironment (TME), which includes tumor cells, fibroblasts, endothelial cells, and inflammatory cells as well as the products of these cells, such as cytokines, extracellular matrix, growth factors, enzymes, and cellular metabolites [5]. Macrophages are the most abundant resident cells in the TME [6]. Depending on different microenvironmental stimuli, macrophages can differentiate into classically activated macrophage (M1 type) or alternatively activated macrophage (M2 type) phenotypes [7]. Toll-like receptor ligands such as IFN-γ and LPS activate M1 macrophages, which can exhibit potent antimicrobial properties and promote inflammatory responses. In contrast, macrophages are polarized into an M2 phenotype when stimulated with Th2 cytokines such as IL-4 or IL-13, and M2 macrophages can facilitate tissue repair and suppress immune responses [8].
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that is downregulated in various cancers [9]. By dephosphorylating phosphatidylinositol-3,4,5-trisphosphate (PIP3) to phosphatidylinositol-(3,4)-biphosphate (PIP2), PTEN antagonizes the activity of PI3K, which eventually inhibits the PI3K/AKT signaling pathway [10]. Moreover, PTEN plays an important role in cellular processes such as cell proliferation, apoptosis, migration, and angiogenesis [11], and dysregulation of PTEN has been detected in numerous types of cancers, including gliomas [12], breast cancer [13], and bladder cancer [14].
MicroRNAs (miRNAs) are a class of endogenous small noncoding RNAs of approximately 22 nucleotides in length that regulate gene expression post-transcriptionally [15]. MicroRNAs can negatively regulate target gene expression by directing mRNA degradation or translational repression, eventually leading to a reduction in protein levels [16]. Many cellular pathways and functions, including cell metabolism, differentiation, and apoptosis, can be regulated by miRNAs, so dysregulation of microRNAs can lead to multiple human diseases as well as obesity, senile dementia, and cancers [17]. Recent studies have discovered that abnormal miRNA expression is involved in the tumorigenesis, progression, and metastasis of gliomas [18]. miR-32 has been considered an oncogene in castration-resistant prostate cancer, gastric carcinoma and colorectal carcinoma [19,20,21]. However, the effect of miR-32 on glioma and its specific mechanism have not been studied, which may provide new insight into the treatment of glioma.
In this study, we illustrated that miR-32 targets PTEN, inducing the polarization of macrophages to an M2 phenotype through the PI3K/AKT pathway, in turn affecting glioma proliferation, apoptosis, and migration.
Methods
Cell culture
The human glioblastoma cell line U87 and human monocytic cell line THP1 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). The THP1 cell line was cultured in Roswell Park Memorial Institute-1640 medium (RPMI-1640; Gibco, Waltham, MA, USA) supplemented with 1% penicillin–streptomycin (1% P/S) and 10% fetal bovine serum (FBS; Gibco, Waltham, MA, USA). The U87 cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Waltham, MA, USA) with 1% P/S and 10% FBS. Cells were cultured at 37 °C in a humidified incubator containing 5% CO2. Cells and the culture supernatant were preserved for future analysis. To induce M2 macrophage polarization, THP1 cells (1 × 106) were incubated in 50% complete medium plus 50% U87-conditioned medium.
Western blotting
U87 or THP1 cells were lysed using RIPA lysis buffer (Thermo Scientific, Waltham, MA, USA). The concentration of proteins was determined by using a BCA Protein Assay Kit (Beyotime, China). Total proteins (30 μg) were loaded into 10% SDS-PAGE gels, probed with mouse or rabbit mAbs against CD163 (ab182422, 1:1000), CD204 (ab123946, 1:1000), CD206 (ab64693, 1:1500), PTEN (ab32199, 1:1500), PI3K (ab151549, 1:1000), p-AKT (ab81283, 1:1000), AKT (ab8805, 1:1000), Bad (ab32445, 1:1000), Bcl2 (ab32124, 1:1000), active-caspase-9 (ab2324, 1:1000), and β-actin (ab8224, 1:1000), and then incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000; ZSGB-BIO, Beijing, China). The primary antibodies were obtained from Abcam (CAM, UK). β-actin was measured as a loading control. Images were obtained by using a transmission scanner employing actin protein levels as an internal control, and relative quantitative analysis was carried out based on the image band density ratio with ImageJ software (NIH, Bethesda, MD, USA).
ELISA
The THP1 culture supernatant was used for ELISAs. The expression levels of CCL18, CCL22, CXCL2, IL-10, VEGF and TGFβ1 were detected using a CCL18 ELISA kit (ab100620), CCL22 ELISA kit (ab100591), CXCL2 ELISA kit (ab184862), IL-10 ELISA kit (ab100549), VEGF ELISA kit (ab100662) and TGF-β1 ELISA kit (ab100647) according to the manufacturer’s protocols. These ELISA kits were purchased from Abcam (CAM, UK).
Real-time PCR
The total RNAs of THP1 cells were purified and enriched with a TRIzol RNA Isolation Kit (Thermo). miRNAs were extended by stem-loop methods, and total RNA reverse transcription was performed with the TransScript First-Strand cDNA Synthesis Kit (Thermo). After reverse transcription, quantitative real-time PCR was performed to analyze the levels of miRNA and mRNA transcripts using specific primers and the QuantiTect SYBR-Green PCR kit (Qiagen, Duesseldorf, Germany). β-actin and U6 small RNA levels were used to normalize the mRNA and miRNA measurements. The expression levels of miR-32 and PTEN were presented according to the \(2^{{ - \Delta \Delta C_{T} }}\) method. The primer sequences are as follows: miR-32-Forward, 5′-GCGGCGTATTGCACATTACT-3′, and miR-32-Reverse, 5′-TCGTATCCA GTGCAGGGT C-3′; PTEN-Forward, 5′-CGGCAGCATCAAATGTTTCAG-3′, and PTEN-Reverse 5′-AACTGGCAGGTAGAAGGCAACTC-3′; GAPDH-Forward, 5′-ATGTCGTGGAGTCTACTGGC-3′, and GAPDH-Reverse, 5′-TGACCTTGCCCACAGCCTTG-3′; U6-Forward, 5′-GCGCGTCGTGAAGCGTTC-3′, and U6-Reverse, 5′-GTGCAGG GTCCGAGGT-3′.
Luciferase reporter assays
To construct luciferase reporter vectors, the PTEN 3′UTR fragment containing putative miR-32 binding sites was amplified using PCR and inserted into the p-MIR-reporter plasmid (Ambion, Austin TX, USA); this construct is referred to as PTEN WT (wild type). Site-directed mutagenesis of the miR-32 target site in the PTEN 3′UTR was performed, and the product was inserted into the p-MIR-reporter plasmid to establish PTEN MT (mutant type). The plasmids were validated by DNA sequencing. THP1 cells were cultured in 24-well plates (5 × 104 cells/well), and each well was cotransfected with PTEN WT or PTEN MT and a miR-23 mimic or inhibitor or NC (negative control), together with a β-galactosidase (β-gal) expression plasmid (Ambion, Austin, TX, USA), using Lipofectamine™ 2000. The β-gal expression plasmid was detected as a transfection control. Cells were collected after 48 h of transfection, and the cells were assayed using a luciferase assay kit (Active Motif, Carlsbad, CA, USA).
Cell transfection
The miR-32 mimic, miR-32 inhibitor, miR-32 NC, pcDNA3.1-PTEN, and pcDNA3.1 were all purchased from GenePharma (Shanghai, China). THP1 cells were transfected with the miR-32 mimic, miR-32 inhibitor, miR-32 NC, miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN, or miR-32 NC + pcDNA3.1. Transfection was conducted using Lipofectamine 2000 (Thermo) following the manufacturer’s protocols. After 24 h of transfection, the cells were incubated with the U87 cell culture supernatant for 48 h, and the supernatant of culture medium was then harvested for further experiments.
Cell Counting Kit-8 assay
For this assay, 1000 U87 cells were seeded in a 96-well plate and incubated in 50% complete medium plus 50% THP1 conditioned medium. After the cells had been cultured for 0, 24, 48, or 72 h-, cell proliferation was measured by the Cell Counting Kit-8 (CCK-8; Dojindo, Tokyo, Japan) assay on the basis of the manufacturer’s instructions.
Flow cytometry
The apoptosis assay was performed with an Annexin-V-FITC/PI apoptosis detection kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s manual. THP1 cells after transfection or U87 cells cocultured with THP1-conditioned medium for 7 days were washed by twice with PBS (Phosphate Buffered Saline) buffer. Then, the cells were resuspended in 1 × binding buffer at a concentration of 1 × 106 cells/ml, and 5 μl of Annexin-V-FITC conjugate and 10 μl of propidium iodide (PI) solution were added to each 500 μl cell suspension. Cells were stained by Annexin-V-FITC/PI for 10 min at room temperature. Stained samples were analyzed using a MoFlo XDP flow cytometer (Beckman Coulter, Brea, CA, USA), and the apoptosis rate was determined using FlowJo software (Tree Star, Ashland, OR, USA).
Colony forming assay
For this assay, 1000 U87 cells were plated in six-well plates and cocultured with THP1-conditioned medium for 2 weeks. After 2 weeks of incubation, the cells were washed twice with PBS, then stained with crystal violet, and the numbers of colonies per well were counted. Colonies that exceeded 75 μm in diameter or contained at least 50 cells were counted as 1 positive colony.
Wound healing assay
A wound healing assay was performed to assess the U87 cell migration capacity. Briefly, U87 cells (1 × 106/well) were seeded in six-well plates and cultured in 50% complete medium plus 50% THP1-conditioned medium. Upon reaching confluency, the cell layer was scratched using a sterile plastic tip, then washed twice with serum-free culture medium and cultured again for 48 h with serum-reduced culture medium containing 1% FBS. Images of the plates were taken using a microscope.
Transwell assay
The cell invasion ability was measured using Transwell chambers (8-μm pore; BD Biosciences). U87 cells (5 × 104 cells) suspended in 500 μl of serum-free culture medium were added to the upper chamber. The lower chamber was seeded with THP1 cells subjected to different treatments, including miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN, and miR-32 NC + pcDNA3.1 (NC). In addition, THP1 cells from all groups were cultured with conditioned supernatant mixed with complete medium at a ratio of 1:1. After 48 h, the non-invading cells on the upper surface were separated, and the cells that had invaded to the bottom of the membrane were fixed with methanol and stained with 0.1% crystal violet, and digital image acquisition was performed after air drying. The number of invasive cells was counted by using a microscope.
Statistical analysis
All of the statistical analyses were carried out by using GraphPad Prism 6. Values were presented as the mean ± SD with of least three independent experiments in triplicate. Significance was determined using Student’s t test, and a p value lower than 0.05 was considered statistically significant.
Results
The glioma microenvironment promotes M2 macrophage polarization of THP1 cells along with upregulation of miR-32 and downregulation of PTEN
To mimic the glioma microenvironment, we cocultured the human monocytic cell line THP1 with the human glioblastoma cell line U87 culture supernatant (referred to as the induced group). Figure 1a and b show that the protein expression levels of M2 macrophage-specific markers in both the THP1 cell lysate (CD206, CD204, CD163) and THP1 culture supernatant (CCL18, CCL22, CXCL2, IL-10, VEGF, TGF beta1) were significantly increased in the induced group in comparison with the control group. These results suggested that the U87 culture supernatant can polarize THP1 cells to an M2 macrophage phenotype. In addition, we observed that the glioma microenvironment changed miRNA expression patterns in THP1 cells. Our data indicate that the miR-32 expression level in THP1 cells in the induced group was significantly upregulated relative to that in the control group (Fig. 1c). Simultaneously, we confirmed that the mRNA and protein levels of PTEN were significantly downregulated (Fig. 1d, e). The results showed that miR-32 and PTEN may be involved in THP1 macrophage polarization.

THP1 cells were cocultured with the U87 culture supernatant for 48 h to induce M2 macrophage polarization. a The protein levels of CD206, CD204 and CD16 in THP1 cells were determined by western blotting. b The activity levels of CCL18, CCL22, CXCL2, IL-10, VEGF and TGFbeta1 in the THP1 culture supernatant were measured by ELISA. The expression of miR-32 in THP1 cells was significantly upregulated, while the expression of PTEN was significantly downregulated. c, d The RNA expression of miR-32 and PTEN in THP1 cells was detected by RT-qPCR. e The expression of PTEN was examined by western blotting in THP1 cells. Data are shown as the mean ± SD, n = 3. Experiments were performed in triplicate. *p < 0.05, **p < 0.01 compared with the control group
miR-32 can regulate PTEN gene expression in THP1 cells
We searched for putative miR-32 binding sequences in the PTEN 3′UTR using the miRNA target analysis tool TargetScan (Fig. 2a). To confirm the target relation of miR-32 and PTEN, a fragment of the 3′-UTR of PTEN mRNA containing the putative or mutated miR-32 binding sequence was cloned into a luciferase reporter plasmid. Luciferase activity assays showed that the miR-32 mimic significantly inhibited luciferase activity in the WT group in contrast to the MT group, while the miR-32 inhibitor significantly promoted luciferase activity in the WT group in THP1 cells (Fig. 2b). We further transfected THP1 cells with miR-32 NC or the miR-32 mimic or inhibitor. A similar result was observed, where overexpression of miR-32 significantly depressed both the mRNA and protein expression of PTEN, and inhibition of miR-32 increased both the mRNA and protein expression of PTEN in contrast to the NC group (Fig. 2c, d). These data suggest that PTEN is a target of miR-32 and that miR-32 can regulate PTEN gene expression in THP1 cells.

miR-32 targets PTEN and inhibits PTEN expression in THP1 cells. a Putative miR-32 binding sequences within the 3′UTR of PTEN. HEK293 cells were cotransfected with the luciferase reporter plasmid carrying the wild-type (WT) or mutant (MUT) PTEN 3′UTR and miR-32 negative control mimics or inhibitors. At 48 h after transfection, b luciferase activity was determined by luciferase assays. For further verification, THP1 cells were transfected with miR-32 NC or the miR-32 mimic or inhibitor. RNA expression levels of c miR-32 and d PTEN in THP1 cells were detected by RT-qPCR; e the protein expression of PTEN was measured by western blotting. Data are shown as the mean ± SD, n = 3. Experiments were performed in triplicate. *p < 0.05, **p < 0.01 compared with the NC group
miR-32 promotes M2 macrophage polarization by inhibiting PTEN expression in THP1 cells
To examine whether miR-32 affects the M2 phenotypic polarization of macrophages owing to PTEN inhibition, THP1 cells were cotransfected with the miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN, or miR-32 NC + pcDNA3.1. At 24 h after transfection, the THP1 cells were cocultured with U87-conditioned medium for 48 h. Figure 3A shows that transfection of the miR-32 mimic significantly increased the miR-32 expression level, while transfection of pcDNA3.1-PTEN significantly decreased the miR-32 expression level compared with the NC group. The miR-32 mimic significantly reduced both the mRNA and protein expression levels of PTEN; conversely, pcDNA3.1-PTEN significantly promoted the mRNA and protein expression of PTEN and effectively restored the effects of the miR-32 mimic (Fig. 3b). Simultaneously, we detected M2 macrophage-specific markers in both THP1 cells (CD206, CD204, CD163) and the THP1 culture supernatant (CCL18, CCL22, CXCL2, IL-10, VEGF, TGF beta1). The data suggested that the miR-32 mimic significantly upregulated while pcDNA3.1-PTEN significantly downregulated the expression of these M2 markers compared with the NC group, and pcDNA3.1-PTEN also obviously rescued the inhibitory effect of the miR-32 mimic (Fig. 3c, d). Together, the results showed that miR-32 inhibits PTEN expression, thus promoting M2 transformation in THP1 cells.

miR-32 promotes the transformation of THP1 cells to M2 macrophages by targeting PTEN. THP1 cells were cotransfected with the miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN or miR-32 NC + pcDNA3.1. At 24 h after transfection, the THP1 cells were cocultured with U87-conditioned medium for 48 h. The RNA expression levels of a miR-32 and b PTEN in THP1 cells were detected by RT-qPCR. c The protein expression levels of CD206, CD204, CD16, PTEN in THP1 cells were determined by western blotting. d The activity levels of CCL18, CCL22, CXCL2, IL-10, VEGF and TGFbeta1 in the THP1 culture medium supernatant were measured by ELISA. Data are shown as the mean ± SD, n = 3. Experiments were performed in triplicate. NC included empty pcDNA3.1 and the miR-32 negative control. **p < 0.01 compared with the NC group. ##p < 0.01 compared with the mimic group
miR-32 promotes M2 macrophage polarization of THP1 cells by targeting PTEN through the PI3K/AKT signaling pathway
Since PTEN is closely related to cell apoptosis via activating the PI3K/AKT signaling pathway, we also used flow cytometry to detect cell apoptosis in every group. The miR-32 mimic significantly suppressed THP1 cell apoptosis compared with the NC group. In contrast, pcDNA3.1-PTEN significantly promoted cell apoptosis and reversed the apoptosis resistance effect of the miR-32 mimic (Fig. 4a, b). To further obverse the potential regulatory mechanisms of M2 transformation and macrophage apoptosis, we detected the expression levels of the PI3K/AKT signaling pathway proteins PI3K, AKT, and p-AKT and the downstream apoptosis-related proteins Bad, Bcl2, and active-caspase-9. We found that overexpression of miR-32 significantly enhanced PI3K and p-AKT expression compared with the NC group, thus activating the PI3K/AKT signaling pathway, causing the pro-apoptotic proteins Bad and active-caspase-9 to be significantly downregulated and the apoptosis inhibitory protein Bcl2 to be significantly upregulated. In contrast, after the transfection of pcDNA3.1-PTEN, the expression of PI3K and p-AKT was significantly decreased compared with the NC group, and repression of the PI3K/AKT signaling pathway caused significant overexpression of the pro-apoptotic protein Bad and downregulation of the apoptosis inhibitory protein Bcl2 (Fig. 4c, d). Our results showed that miR-32 activates the PI3K/AKT signaling pathway by downregulating PTEN, after which the activated PI3K/AKT signaling pathway induces M2 macrophage polarization and regulates the expression of the apoptosis-related proteins Bad, Bcl2, and active-caspase-9, thereby inhibiting the apoptosis of THP1 cells.

PTEN can reverse the inhibitory effect of miR-32 on THP1 cell apoptosis. THP1 cells were cotransfected with the miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN or miR-32 NC + pcDNA3.1. At 24 h after transfection, THP1 cells were incubated with U87-conditioned medium for 48 h. a, b THP1 cell apoptosis was analyzed by flow cytometry. c, d The expression levels of PTEN, PI3K, p-AKT, AKT Bad, Bcl2 and active-caspase-9 were measured by western blotting. **p < 0.01 compared with the NC group. ##p < 0.01 compared with the mimic group
Downregulation of PTEN by miR-32 in THP1 cells promotes proliferation and inhibits apoptosis in U87 cells
To prove that miR-32 expressed in macrophages plays a pivotal role in the glioma microenvironment and can alter the proliferation and apoptosis capacity of glioma cells, we performed the CCK-8 assay, colony forming assay and apoptosis assay. U87 cells were cocultured with conditioned medium from transfected THP1 cells (miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN, or miR-32 NC + pcDNA3.1). The results of the CCK-8 assay indicated that the miR-32 mimic promoted the growth of U87 cells and that pcDNA3.1-PTEN inhibited the growth of U87 cells compared with NC. When THP1 cells were transfected with the mimic and pcDNA3.1-PTEN simultaneously, the cell proliferative index of U87 cells exhibited significant reversal compared with the group transfected with the miR-32 mimic alone (Fig. 5a). Flow cytometry was used to detect the apoptosis of U87 cells in each group. It was found that the miR-32 mimic significantly inhibited the apoptosis of U87 cells and that PTEN significantly promoted the apoptosis of U87 cells (Fig. 5b, c). Similarly, the miR-32 mimic promoted the clone formation ability of U87 cells, while pcDNA3.1-PTEN inhibited the clone formation ability of U87 cells compared with NC, and overexpression of PTEN reversed the effect of the miR-32 mimic (Fig. 5d). In conclusion, miR-32 downregulated PTEN expression in THP1 cells, promoted the proliferation of U87 cells and inhibited the apoptosis of U87 cells.

miR-32 promotes the proliferation and suppresses the apoptosis of U87 cells by targeting PTEN. U87 cells were cocultured with THP1-conditioned medium subjected to four different transfection treatments (miR-32 mimic + pcDNA3.1-PTEN, miR-32 mimic + pcDNA3.1, miR-32 NC + pcDNA3.1-PTEN or miR-32 NC + pcDNA3.1) for 7 days. a The CCK-8 assay was used to detect U87 cell proliferation at 0, 24, 48 and 72 h after incubation. b, c U87 cell apoptosis was analyzed by flow cytometry. (D) Colony forming assays revealed the cell proliferation and growth of each U87 cell group. *p < 0.05, **p < 0.01 compared with the NC group. ##p < 0.01 compared with the mimic group
Downregulation of PTEN by miR-32 in THP1 cells promotes the migration and invasion of U87 cells
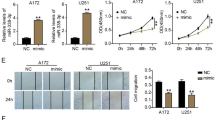
We further performed Transwell assays and wound healing assays to assess the effect of miR-32 expression in THP1 cells on glioma cell invasion and migration abilities. We found that the miR-32 mimic significantly increased the number of migrating cancer cells in U87 cells compared with NC. Overexpression PTEN in THP1 cells significantly reduced the number of migrating cancer cells in U87 cells versus the NC group. In addition, we cotransfected the miR-32 mimic with pcDNA3.1-PTEN, and the results showed that pcDNA3.1-PTEN reversed the effect of the miR-32 mimic compared to the mimic group (Fig. 6a, b). In the wound healing assay, U87 cells were cultured in 50% complete medium plus 50% THP1-conditioned medium. We found that overexpression of the miR-32 mimic significantly enhanced the wound healing rate of U87 cells versus the NC group. PTEN overexpression not only significantly reduced the wound healing rate compared with the NC group but also reversed the effect of the miR-32 mimic compared to the mimic group (Fig. 6c, d). These results implied that miR-32 downregulated PTEN expression in THP1 cells promoted migration and invasion of U87 cells.

miR-32 promotes the migration and invasion ability of U87 cells by targeting PTEN. The U87 cells were added to the upper Transwell chamber, and transfected THP1-conditioned medium mixed with complete medium at a ratio of 1:1 was added to the lower chamber. a, b The Transwell assay was used to detect the changes in the U87 cell invasion capacity. U87 cells were seeded in six-well plates and cultured in THP1-conditioned medium mixed with complete medium at a ratio of 1:1. c, d The wound healing assay was used to detect the changes in the cell migration capacity. *p < 0.05, **p < 0.01 compared with the NC group. ##p < 0.01 compared with the mimic group
Discussion
In the present study, we found that miR-32 negatively regulated PTEN gene expression and consequently activated the PI3K/AKT pathway to inhibit macrophage apoptosis and promote M2 macrophage polarization. Ultimately, overexpression of miR-32 facilities M2 macrophage polarization and promotes glioma proliferation, migration and motility in the glioma microenvironment.
The tumor-associated macrophages (TAMs) are present in the tumor microenvironment and exert key effects on tumor-related inflammation, immunity, matrix remodeling, and metastasis [22]. As a critical component of the tumor microenvironment in gliomas, TAMs are enriched in gliomas and play key roles in glioma proliferation, survival and migration [23]. The accumulation and activity of TAM markers have been reported to correlate with glioma progression and tumor grade and may be applied to predict poor survival of glioblastoma patients [24, 25]. Sufficient studies have revealed that TAMs can be functionally classified into tumor-suppressive macrophages (M1 type) and tumor-supportive (M2 type) macrophages [26]. M1 TAMs present pro-inflammatory activity, thus hampering tumor growth and prolonging patient survival [27]. In contrast, M2 TAMs are associated with a poor prognosis, are generally immune-suppressive and contribute to tumor growth. TAMs are more similar to M2 than M1 macrophages overall [28]. TAM-targeted therapy has been shown to potently relieve glioma progression in animals, indicating that TAMs are potential targets for glioma treatment [29, 30]. In our study, we also found that M2 macrophage polarization promotes glioma proliferation, migration, and motility in the glioma microenvironment.
Recently, many studies have revealed the relationship between PTEN and macrophage polarization. Li et al. showed that PTEN suppressed the transformation of M2 macrophages in the tumor microenvironment [31], and PTEN in coordination with NHERF-1 inhibits M2 macrophage phenotype polarization in the tumor microenvironment by decreasing CCL2 and VEGF-A levels [32]. In addition, Cheng et al. found that PTEN mediates the M2 polarization of macrophages via activation of PI3K/Akt/STAT6 signaling in liver fibrosis [33]. Furthermore, the GSK3β/PTEN/Akt axis can modulate microglia/macrophage polarization in white matter injury [34]. Here, we obtained similar results showing that downregulation of PTEN promoted M2 macrophage polarization.
As a main downstream pathway of PTEN, the PI3K/AKT pathway plays an important role in various cellular processes, including cell survival, proliferation, migration, and differentiation [35]. PI3K can catalyze the production of PIP3, subsequently activating protein kinase AKT [36]. The AKT family consists of three serine/threonine protein kinases (Akt1, Akt2, and Akt3) and has been shown to regulate apoptosis-related proteins such as Bcl-2, Bax, Bad, and caspase-9, thus promoting survival and blocking apoptosis in some cancer cells [37, 38]. Recently, Zhang et al. observed that a PI3K/Akt pathway inhibitor can suppress M2 macrophage polarization by downregulating paxillin, thereby suppressing colon cancer cell proliferation and migration [39]. Additionally, luteolin has been found to regulate macrophage polarization via the PI3K/Akt Pathway to inhibit apoptosis stimulated by Angiotensin II [40]. Furthermore, Tian et al. confirmed that adenosine 2B receptor activation significantly increased the number of M2 macrophages and decreased the number of M1 macrophages via the PI3K/Akt pathway, thus reducing myocardial ischemia/reperfusion (IR) injury [41]. In accordance with these findings, we found that miR-32 activated the PI3K/AKT pathway by targeting PTEN, thereby promoting M2 macrophage polarization and inhibiting macrophage apoptosis.
In conclusion, miR-32 promotes M2 macrophage polarization by targeting PTEN though the PI3K/AKT pathway and further promoting the proliferation, migration, and invasion and inhibiting the apoptosis of human glioma cells in the tumor microenvironment. Our results illustrate the association of miR-32, PTEN, and the PI3K/Akt pathway, which provides new insight into prognostic and therapeutic strategies for gliomas.
References
Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS (2016) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro Oncol 18:v1–v75. https://doi.org/10.1093/neuonc/now207
Lapointe S, Perry A, Butowski NA (2018) Primary brain tumours in adults. Lancet 392:432–446. https://doi.org/10.1016/S0140-6736(18)30990-5
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016) WHO classification of tumours of the central nervous system, revised 4th edn. World Health Organization, Geneva
Guo M, Zhang X, Wang G, Sun J, Jiang Z, Khadarian K, Yu S, Zhao Y, Xie C, Zhang K, Zhu M, Shen H, Lin Z, Jiang C, Shen J, Zheng Y (2015) miR-603 promotes glioma cell growth via Wnt/beta-catenin pathway by inhibiting WIF1 and CTNNBIP1. Cancer Lett 360:76–86. https://doi.org/10.1016/j.canlet.2015.02.003
Ren F, Fan M, Mei J, Wu Y, Liu C, Pu Q, You Z, Liu L (2014) Interferon-gamma and celecoxib inhibit lung-tumor growth through modulating M2/M1 macrophage ratio in the tumor microenvironment. Drug Des Devel Ther 8:1527–1538. https://doi.org/10.2147/DDDT.S66302
Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66:605–612. https://doi.org/10.1158/0008-5472.CAN-05-4005
Bi J, Zeng X, Zhao L, Wei Q, Yu L, Wang X, Yu Z, Cao Y, Shan F, Wei M (2016) miR-181a induces macrophage polarized to M2 phenotype and promotes M2 macrophage-mediated tumor cell metastasis by targeting KLF6 and C/EBPalpha. Mol Ther Nucleic Acids 5:e368. https://doi.org/10.1038/mtna.2016.71
Wang Z, Brandt S, Medeiros A, Wang S, Wu H, Dent A, Serezani CH (2015) MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PLoS ONE 10:e0115855. https://doi.org/10.1371/journal.pone.0115855
Song H, Zhang Y, Liu N, Wan C, Zhang D, Zhao S, Kong Y, Yuan L (2016) miR-92b regulates glioma cells proliferation, migration, invasion, and apoptosis via PTEN/Akt signaling pathway. J Physiol Biochem 72:201–211. https://doi.org/10.1007/s13105-016-0470-z
Zhou K, Zhang T, Fan Y, Serick, Du G, Wu P, Geng D (2016) MicroRNA-106b promotes pituitary tumor cell proliferation and invasion through PI3K/AKT signaling pathway by targeting PTEN. Tumour Biol 37:13469–13477. https://doi.org/10.1007/s13277-016-5155-2
Li KK, Xia T, Ma FM, Zhang R, Mao Y, Wang Y, Zhou L, Lau KM, Ng HK (2015) miR-106b is overexpressed in medulloblastomas and interacts directly with PTEN. Neuropathol Appl Neurobiol 41:145–164. https://doi.org/10.1111/nan.12169
Elhag R, Mazzio EA, Soliman KF (2015) The effect of silibinin in enhancing toxicity of temozolomide and etoposide in p53 and PTEN-mutated resistant glioma cell lines. Anticancer Res 35:1263–1269
Zhou W, Shi G, Zhang Q, Wu Q, Li B, Zhang Z (2014) MicroRNA-20b promotes cell growth of breast cancer cells partly via targeting phosphatase and tensin homologue (PTEN). Cell Biosci 4:62. https://doi.org/10.1186/2045-3701-4-62
Feng Y, Liu J, Kang Y, He Y, Liang B, Yang P, Yu Z (2014) miR-19a acts as an oncogenic microRNA and is up-regulated in bladder cancer. J Exp Clin Cancer Res 33:67. https://doi.org/10.1186/PREACCEPT-9242556491295527
Cai G, Qiao S, Chen K (2015) Suppression of miR-221 inhibits glioma cells proliferation and invasion via targeting SEMA3B. Biol Res 48:37. https://doi.org/10.1186/s40659-015-0030-y
Su Y, He Q, Deng L, Wang J, Liu Q, Wang D, Huang Q, Li G (2014) MiR-200a impairs glioma cell growth, migration, and invasion by targeting SIM2-s. NeuroReport 25:12–17. https://doi.org/10.1097/WNR.0000000000000032
Mo YY (2012) MicroRNA regulatory networks and human disease. Cell Mol Life Sci 69:3529–3531. https://doi.org/10.1007/s00018-012-1123-1
Yin F, Zhang JN, Wang SW, Zhou CH, Zhao MM, Fan WH, Fan M, Liu S (2015) MiR-125a-3p regulates glioma apoptosis and invasion by regulating Nrg1. PLoS ONE 10:e0116759. https://doi.org/10.1371/journal.pone.0116759
Yan C, Yu J, Liu Y, Kang W, Ma Z, Zhou L (2015) MiR-32 promotes gastric carcinoma tumorigenesis by targeting Kruppel-like factor 4. Biochem Biophys Res Commun 467:913–920. https://doi.org/10.1016/j.bbrc.2015.10.044
Wu W, Yang J, Feng X, Wang H, Ye S, Yang P, Tan W, Wei G, Zhou Y (2013) MicroRNA-32 (miR-32) regulates phosphatase and tensin homologue (PTEN) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol Cancer 12:30. https://doi.org/10.1186/1476-4598-12-30
Jalava SE, Urbanucci A, Latonen L, Waltering KK, Sahu B, Janne OA, Seppala J, Lahdesmaki H, Tammela TL, Visakorpi T (2012) Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene 31:4460–4471. https://doi.org/10.1038/onc.2011.624
Ma ZG, Yuan YP, Wu HM, Zhang X, Tang QZ (2018) Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci 14:1645–1657. https://doi.org/10.7150/ijbs.28103
Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19:20–27. https://doi.org/10.1038/nn.4185
Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, Ancukiewicz M, Plotkin SR, Chi AS, Gerstner ER, Eichler AF, Dietrich J, Stemmer-Rachamimov AO, Batchelor TT, Jain RK (2013) Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol 15:1079–1087. https://doi.org/10.1093/neuonc/not082
Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, Heber-Katz E, Curtis MT, Cotzia P, Hooper DC (2013) Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res 19:3776–3786. https://doi.org/10.1158/1078-0432.CCR-12-1940
Ostuni R, Kratochvill F, Murray PJ, Natoli G (2015) Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 36:229–239. https://doi.org/10.1016/j.it.2015.02.004
Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, Zhang L, Chen L, Lan X, Zhang XC, Zhou K, Liu Q, Long H, Fu TW, Zhang XN, Cao MF, Huang Z, Fang X, Wang X, Feng H, Yao XH, Yu SC, Cui YH, Zhang X, Rich JN, Bao S, Bian XW (2017) Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun 8:15080. https://doi.org/10.1038/ncomms15080
Zhu C, Kros JM, van der Weiden M, Zheng P, Cheng C, Mustafa DA (2017) Expression site of P2RY12 in residential microglial cells in astrocytomas correlates with M1 and M2 marker expression and tumor grade. Acta Neuropathol Commun 5:4. https://doi.org/10.1186/s40478-016-0405-5
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN, Bao S (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17:170–182. https://doi.org/10.1038/ncb3090
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19:1264–1272. https://doi.org/10.1038/nm.3337
Li N, Qin JF, Han X, Jin FJ, Zhang JH, Lan L, Wang Y (2018) miR-21a negatively modulates tumor suppressor genes PTEN and miR-200c and further promotes the transformation of M2 macrophages. Immunol Cell Biol 96:68–80. https://doi.org/10.1111/imcb.1016
Li N, Qin J, Lan L, Zhang H, Liu F, Wu Z, Ni H, Wang Y (2015) PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol Ther 16:297–306. https://doi.org/10.1080/15384047.2014.1002353
Cheng Y, Tian Y, Xia J, Wu X, Yang Y, Li X, Huang C, Meng X, Ma T, Li J (2017) The role of PTEN in regulation of hepatic macrophages activation and function in progression and reversal of liver fibrosis. Toxicol Appl Pharmacol 317:51–62. https://doi.org/10.1016/j.taap.2017.01.005
Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW, Tang B, Wang Y, Gao Y, Zheng P, Bennett MV, Chen J (2015) HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci USA 112:2853–2858. https://doi.org/10.1073/pnas.1501441112
Zhou BH, Tan PP, Jia LS, Zhao WP, Wang JC, Wang HW (2018) PI3K/AKT signaling pathway involvement in fluoride-induced apoptosis in C2C12cells. Chemosphere 199:297–302. https://doi.org/10.1016/j.chemosphere.2018.02.057
Ying J, Xu Q, Liu B, Zhang G, Chen L, Pan H (2015) The expression of the PI3K/AKT/mTOR pathway in gastric cancer and its role in gastric cancer prognosis. Onco Targets Ther 8:2427–2433. https://doi.org/10.2147/OTT.S88592
Zhu Z, Sun H, Ma G, Wang Z, Li E, Liu Y, Liu Y (2012) Bufalin induces lung cancer cell apoptosis via the inhibition of PI3K/Akt pathway. Int J Mol Sci 13:2025–2035. https://doi.org/10.3390/ijms13022025
Li D, Qu X, Hou K, Zhang Y, Dong Q, Teng Y, Zhang J, Liu Y (2009) PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs 20:59–64. https://doi.org/10.1097/CAD.0b013e3283160fd6
Zhang LL, Zhang LF, Shi YB (2018) Down-regulated paxillin suppresses cell proliferation and invasion by inhibiting M2 macrophage polarization in colon cancer. Biol Chem 399:1285–1295. https://doi.org/10.1515/hsz-2018-0002
Jiang Q, Pan D, Yang Y, Hu Y, Fang L, Shang P, Xia Y, Li D (2018) Luteolin regulates macrophage polarization via the PI3K/Akt pathway to inhibit the apoptosis stimulated by angiotensin II. Curr Pharm Biotechnol 19:428–437. https://doi.org/10.2174/1389201019666180629143251
Tian Y, Piras BA, Kron IL, French BA, Yang Z (2015) Adenosine 2B receptor activation reduces myocardial reperfusion injury by promoting anti-inflammatory macrophages differentiation via PI3K/Akt pathway. Oxid Med Cell Longev 2015:585297. https://doi.org/10.1155/2015/585297
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Research Involving Human Participants and/or Animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bao, L., Li, X. MicroRNA-32 targeting PTEN enhances M2 macrophage polarization in the glioma microenvironment and further promotes the progression of glioma. Mol Cell Biochem 460, 67–79 (2019). https://doi.org/10.1007/s11010-019-03571-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-019-03571-2