
Abstract
Oxidative stress and neuroinflammation contribute significantly to the development and progression of diabetic retinopathy. Fenofibrate has received great attention as it benefits diabetic patients by reducing retinal laser requirement. Nuclear factor erythroid-2-related factor 2 (Nrf2) is a master regulator of anti-oxidative defense. Activation of nucleotide binding domain, leucine-rich repeat-containing receptor (NLR), pyrin domain-containing 3 (NLRP3) inflammasome plays a pivotal role in neuroinflammation. The purpose of this study is to determine whether fenofibrate protects retinas from oxidative damage and neuroinflammation via modulating the Nrf2 pathway and blocking NLRP3 inflammasome activation during diabetes. Diabetes is induced by intraperitoneal injection of streptozotocin in mice. Fenofibrate was given to mice in rodent chow. Upregulation of Nrf2 and NLRP3 inflammasome, enhanced ROS formation, and increased leukostasis and vascular leakage were observed in diabetic mouse retinas. Notably, Nrf2 and Caspase-1 were mainly colocalized with glutamine synthetase, one of the Mȕller cell markers. Fenofibrate further increased the expression of Nrf2 and its target gene NQO-1 and HO-1 and reduced ROS formation in diabetic retinas. In addition, retinal expression of NLRP3, Caspase-1 p20, IL-1β p17, and ICAM-1 were dramatically increased in vehicle-treated diabetic mice, which were abolished by fenofibrate intervention. Moreover, fenofibrate treatment also attenuated diabetes-induced retinal leukostasis and vascular leakage in mice. Taken together, fenofibrate attenuates oxidative stress and neuroinflammation in diabetic retinas, which is at least partially through modulating Nrf2 expression and NLRP3 inflammasome activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic retinopathy (DR) is a common neurovascular complication of diabetes, which is the most frequent cause of vision loss and blindness among working age population in both developed and developing countries. Multiple risk factors contribute to DR including chronic hyperglycemia, hypertension, and dyslipidemia. Intensive glycemic control has been shown to lower the risk of diabetic complications but it still does not completely prevent the onset and progression of DR. This may be attributed to the complexity of pathogenesis of DR. Thus, clarifying disturbed signaling responsible for diabetes-induced retinal injury will facilitate identification of novel therapeutic targets and further elucidate the potential mechanisms for DR.
Fenofibrate, a peroxisome proliferator-activated receptor-alpha (PPARα) agonist, is a triglyceride-lowing drug and has been widely used at clinic for more than 30 years. Recently, fenofibrate has gained great attention, because of two landmark clinical trials: The Fenofibrate Intervention and Event Lowing in Diabetes (FIELD) and the Action to Control Cardiovascular Risk in Diabetes (ACCORD) independently proved that fenofibrate could delay the progression of DR and benefit DR patients by reducing retinal laser intervention [1, 2]. Emerging evidence suggests that fenofibrate exerts a broad range of beneficial effects on diabetic complications through regulating multiple signaling pathways involved in oxidative stress, inflammation, angiogenesis, and cell survival, which is largely independent of its lipid-lowing property [3].
Oxidative stress has been identified as an “unifying” mechanism of various diabetic complications [4]. Previous studies demonstrated that fenofibrate activated antioxidant pathways in diabetic kidneys and also reduced reactive oxygen species (ROS) in a rodent model of myocardial ischemia [5, 6]. However, the role and mechanism of fenofibrate on anti-oxidative defense of diabetic retinas have not been fully elucidated. Erythroid-2-related factor 2 (Nrf2), a transcription factor, plays a central role in anti-oxidant defense, which binds to the antioxidant response element (ARE) to regulate the transcription of antioxidant and detoxifying enzymes including NAD(P)H quinine dehydrogenase 1 (NQO-1), heme oxygenase-1 (HO-1), glutathione S-transferase (GST), and et al. Activation of Nrf2 function plays a critical protective role against oxidative stress [7]. Loss of Nrf2 has been shown to exacerbate oxidative damage to retinas [8]. Intriguingly, fenofibrate attenuated type 1 diabetic renal damage by up-regulation of Nrf2 expression [5]. However, whether fenofibrate regulates retinal Nrf2 signaling during diabetes remains to be clarified.
DR is a chronic low-grade inflammatory disease. Inflammasome could be activated in response to a diversity of endogenous metabolic stress to induce sterile inflammation without the presence of overt infection [9]. Thus, it plays a pivotal role in the pathophysiology of metabolic disorders, including diabetes, obesity, and insulin resistance. Previous studies demonstrated that the nucleotide binding domain, leucine-rich repeat-containing receptor (NLR), pyrin domain-containing 3 (NLRP3) inflammasome could be triggered by oxidative stress and has been shown to be involved in pathogenesis of DR [10, 11]. NLRP3 inflammasome forms a molecular platform to activate caspase-1, which catalyzes proteolytic process and secretion of mature IL-1β. IL-1β is a potent pro-inflammatory cytokine that initiates inflammation cascade. Fenofibrate has been shown to accelerate diabetic wound healing by improving impaired EPC function via inhibition of NLRP3 inflammasome pathway [12]. However, the role of fenofibrate on activation NLRP3 inflammasome in diabetic retinas remains unexplored.
In the present study, we determined whether fenofibrate ameliorates retinal oxidative damage through modulating Nrf2 pathway and attenuates neuroinflammation via blocking NLRP3 inflammasome activation during diabetes.
Materials and methods
Animal model
Eight-week-old C57BL/6 male mice were obtained from the Model Animal Research Center of Nanjing University (Nanjing, Jiangsu, China). Care, use and treatment of all animals were in strict agreement with the guidelines of the Association for Research in Vision and Ophthalmology Statement for Use of Animals in Ophthalmic and Vision Research and approved by the institutional animal care and use committees in Nanchang University. Diabetes was induced by daily intraperitoneal injections with streptozotocin (Sigma-Aldrich, St. Louis, MO) at 50 mg/kg body weight in 0.1 M sodium citrate (pH 4.5) for five consecutive days as our previous description [13]. Mice injected with sodium citrate alone were used as non-diabetic control. Mice were deemed as diabetic with fasting blood glucose higher than 250 mg/dL. Fenofibrate (Sigma-Aldrich) was incorporated into rodent chow at 300 mg/kg diet (Trophic Animal Feed High-Tech Co., Ltd, Nantong, China) as described elsewhere [14]. Regular rodent chow was given as vehicle diet. At 8 weeks after diabetes onset, mice were randomly assigned into two groups. One group received fenofibrate diet and the other group was given vehicle diet. After 16-week intervention, the mice were utilized for the desired experiments.
CellROX Green Reagent to assess retinal oxidative stress
Mouse eyeballs were immediately embed in OCT (Sakura Finetek) after harvest and slowly frozen with liquid nitrogen. Tissues were sectioned and incubated with CellROX Green Reagent (2.5 μM, diluted in HBSS, Invitrogen) at 37 °C for 30 min according to previous reports [15, 16]. Images were captured with the same setting parameters by Olympus fluorescence microscopy (Germany) and fluorescence intensity of CellROX was analyzed with Image J (NIH, Bethesda, MD). Briefly, retinal region was defined using polygon tools and mean gray value of selected area was measured.
Immunofluorescence staining
Mouse eyes were enucleated and briefly fixed in 4% paraformaldehyde (PFA, Electron Microscopy Science, Hatfield, PA) for 8 min. The cornea and lens were removed and the eyecups were post-fixed in 4% PFA for 15 min. Eyecups were cryoprotected with gradient sucrose and frozen in OCT (Sakura Finetek, Torrance, CA). Cryoblocks were cut into 7-μm sections by a Leica Cryostat (Leica Biosystems, Wetzlar, Germany). Sections were air-dried and then blocked with 0.5% Triton X-100 (Sigma-Aldrich) and 10% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA) in PBS for 30 min. Sections were then incubated overnight at 4 °C with following antibodies: mouse anti-Nrf2 (mab3925, R&D, Minneapolis, MN), mouse-anti-Caspase-1 (AG-20B-0042, AdipoGen, San Diego, CA), mouse-anti-3-nitrotyrosine (3-NT, ab61392, Abcam, Cambridge, MA) and rabbit-anti-glutamine synthetase (GS, ab73593, Abcam) or rabbit-anti-glial fibrillary acidic protein (GFAP, #12389, Cell Signaling Technology, Danvers, MA) and probed with Alex Fluor 488 or 594-conjugated secondary antibodies (Invitrogen, Thermofisher Scientific, Waltham, MA) for 30 min. Slides were coverslipped with VECTASHIELD mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Images were acquired with Olympus Fluorescence Microscopy and fluorescence intensity was quantified by Image J.
Concanavalin A lectin perfusion to measure retinal leukostasis
Mice were anesthetized with 7% Choral Hydrate. Retinal leukocytes adherent to vasculature were quantified by staining with Concanavalin A-lectin as previous described [17]. Briefly, the chest cavity was open and PBS with heparin was perfused through left ventricle and drained via right atrium to remove non-adherent blood cells. Retinal vasculature and adherent leukocytes were labeled with perfusion of Concanavalin A-lectin Alexa Fluor 488 Conjugate (50 μg/ml, diluted in PBS, Invitrogen). Unbound Concanavalin A lectin was flushed out with PBS perfusion. Eyeballs were fixed in 4% PFA for 2 h. The retinas were dissected, flat mounted and photographed under fluorescence microscopy (Olympus). The adherent leukocytes in retinal vasculature were counted.
Evan’s blue-albumin permeability assay to evaluate retinal vascular leakage
Retinal vascular permeability was analyzed as previously described [15]. Evan’s blue was dissolved in PBS (30 mg/ml, Sigma-Aldrich) and was injected into mice via the caudal vein with 31-gauge insulin syringe at a dose of 30 mg/kg. Evan’s blue was allowed to circulate in the mice for 2 h. The mice were transcardially perfused with 0.1 M sodium citrate (pH 4.2) containing 1% PFA and then retinas were dissected. Retinal extravasated Evan’s blue was extracted in 150 μl formamide for 18 h at 70 °C and supernatant was collected after centrifuging at 70,000 rpm for 30 min at 4 °C. Absorbance was measured at 620 nm using spectrophotometer and concentration of Evan’s blue was calculated according to the standard curve of Evan’s blue dissolved in formamide. Retinas were homogenized in PBS and protein concentration was measured by BCA method (Thermofisher Scientific). Results were expressed as microgram of Evan’s blue per milligram of total retinal protein.
Western blot analysis
Retinas were dissected and homogenized in RIPA buffer (Beyotime, Shanghai, China) with protease inhibitors (Roche Applied Science, Mannheim, Germany). Protein concentration was quantified by BCA method (Thermofisher Scientific). Twenty-five micrograms of total proteins were subjected to SDS–PAGE and transferred to nitrocellulose membrane (Merck Millipore, Billerica, MA). The membranes were blotted with rabbit anti-GFAP antibody (#12389, Cell Signaling Technology), mouse anti-NLRP3 antibody (AG-20B-0014, AdipoGen), mouse anti-Caspase-1 antibody (AG-20B-0042, AdipoGen), goat anti-IL-1β antibody (AF-401-NA, R&D), and goat anti-ICAM-1 antibody (sc-1511, Santa-Cruz Biotechnology, Santa Cruz, CA), followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Vector Laboratories). Signals were visualized with SuperSignal West Dura Substrate (ThermoFisher Scientific) using Bio-Imaging System (Syngene, Frederick, MD). The same membrane was reblotted with anti-β-actin antibody (A5441, Sigma-Aldrich) as loading control. The bands were semi-quantified with Genetool (Syngene) by densitometry.
Quantitative RT-PCR
Retinas were harvested and grinded by mortar and pestle in liquid nitrogen. RNA was extracted from retinas with the Trizol method (Thermofisher Scientific) and reverse-transcripted to cDNA using TransScript First-Strand cDNA Synthesis SuperMix (Transgen, Beijing, China). Quantitative RT-PCR reactions were performed in triplicate with SYBR Green Master Mix (Applied Biosystems, Thermofisher Scientific) using Quantitative PCR system (Applied Biosystems). The comparative CT method was used for mRNA quantification. Data were expressed as 2−ΔΔCT for experimental genes normalized to the endogenous control gene. The following primers were used: NQO-1: Forward 5′-GTCTTCTCTGAATGGGCCAG-3′, Reverse 5′-CCAATCAGCGTTCGGTATTA-3′; HO-1: Forward, 5′-CCTTCAAGGCCTCAGACAAA-3′, Reverse 5′-GAGCCTGAATCGAGCAGAAC-3′; RPL19: Forward 5′-TCACAGCCTGTACCTGAAGG-3′, Reverse 5′-TCGTGCTTCCTTGGTCTTAG-3′.
Statistical analysis
All data are presented as mean ± S.D and analyzed using one-way ANOVA with Bonferroni’s post hoc test by Graphpad Prism 5 (GraphPad Software Inc, San Deigo, CA). A p value < 0.05 was considered statistically significant.
Results
Fenofibrate inhibited retinal oxidative stress in diabetic mice
To determine whether the beneficial effect of fenofibrate on diabetic retinas is related to its anti-oxidation property, we evaluated retinal generation of ROS by a fluorogenic probe (CellROX). The results indicate that CellROX fluorescence was enhanced in retinas of diabetic mice compared with non-diabetic controls (Fig. 1a, b). Note that CellROX fluorescence is strongly detected in the inner and the outer segments of photoreceptors in retinas of diabetic mice, suggesting that inner and the outer segments are the major source of retinal ROS formation during diabetes. Fenofibrate significantly reduced diabetes-elicited fluorescence intensity of CellROX in mouse retinas (Fig. 1a, b). Upon oxidative stress, superoxide anion interacts with nitric oxide to generate more powerful oxidant peroxynitrite. Peroxynitrite rapidly reacts with protein tyrosine residues to form 3-NT and leads to cellular protein dysfunction. Next, we examined whether fenofibrate affected peroxynitrate production in diabetic retinas by examining the 3-NT formation with immunofluorescence staining. In parallel with increased ROS generation, diabetic mice exhibited elevated retinal 3-NT immunofluorescence intensity compared with non-diabetic controls (Fig. 1c, d). Moreover, fenofibrate intervention markedly reduced 3-NT formation in diabetic retinas (Fig. 1c, d). Collectively, these data suggested that fenofibrate could attenuate retinal oxidative stress in diabetes by reducing generation of reactive free radicals.

Fenofibrate attenuated retinal oxidative stress in diabetic mice. Retinal oxidative stress was examined by ROS generation (a, b) and 3-NT formation (c, d). a Retinas were fluorescently stained with CellROX for detection of ROS. Fluorescence of CellROX was intensely increased in diabetic retinas with prominent staining of inner and outer segments of photoreceptor (between red lines). Fenofibrate treatment decreased CellROX intensity in the diabetic mouse retinas. Magnification: ×200 and scale bar 100 μm. b Fluorescence intensity of CellROX was semi-quantified by Image J. c Retinas were immunostained with an antibody for 3-NT, a marker of peroxynitrite formation. Diabetic retinas showed increased 3-NT immunofluorescence density. Fenofibrate intervention dramatically reduced 3-NT immunoreactivity in the diabetic mouse retinas. Magnification: ×400 and scale bar 50 μm. d Fluorescence intensity of retinal 3-NT staining was semi-quantified by Image J. n = 4 for each group. **p < 0.01 versus Non-DM and ‡p < 0.01 or †p < 0.05 versus DM
Fenofibrate-induced Nrf2 signaling in retinal Mȕller cells of diabetic mice
Nrf2 is a master regulator of antioxidant response. We next examined whether fenofibrate protected diabetic retinas from oxidative stress through modulating Nrf2 signaling. A double-labeling experiment was performed to determine the cellular distribution of Nrf2 expression in mouse retinas. GS was co-stained as a maker of retinal Mȕller cells. The results demonstrated that Nrf2 was primarily colocalized with GS in Mȕller cell processes across inner plexus layer (IPL) with strong staining at endfeet of Mȕller cells and retinal outer plexus layer (OPL) as shown in Fig. 2a. Fenofibrate treatment further increased Nrf2 immunofluorescence intensity in diabetic mouse retinas (Fig. 2a). These results suggested that fenofibrate upregulated retinal Nrf2 expression during diabetes. To further verify that fenofibrate activated Nrf2 signaling in diabetic retinas, genes that are known to be downstream targets of Nrf2 pathway such as NQO-1 and HO-1 were determined by quantitative RT-PCR. As shown in Fig. 2b, c, the mRNA levels of NQO-1 and HO-1 were significantly elevated in retinas of diabetic mice compared with non-diabetic controls. Consistently, fenofibrate treatment significantly accentuated retinal mRNA level of NQO-1 and HO-1 in diabetic mice compared with those treated with vehicle (Fig. 2b, c). These data indicated that fenofibrate may attenuate retinal ROS generation through promoting activation of Nrf2 signaling.

Fenofibrate activates Nrf2 signaling in diabetic mouse retinas. a Retinal expression and distribution of Nrf2 were determined by immunofluorescence staining. Nrf2 (green) was primarily colocalized with GS (red). GS is a marker that shows Mȕller glial processes extended across all retinal layers from the inner to the outer limiting membranes. Note that retinal expression of Nrf2 was markedly upregulated in the diabetic mice compared with the non-diabetic controls, which was further increased by fenofibrate treatment. Magnification: ×400 and scale bar 50 μm. (b, c) The mRNA levels of Nrf2 downstream genes such as NQO-1 and HO-1 were measured by quantitative-RT-PCR. Diabetes induced significant upregulation of NQO-1 (b) and HO-1 (c) expression in mouse retinas, which was further increased by fenofibrate treatment. n = 4 for each group. **p < 0.01 versus Non-DM and ‡ p < 0.01 versus DM
Fenofibrates attenuated Mȕller cell gliosis in diabetic mice
Reactive gliosis could be triggered in retinas by various insults, which is characterized by increased expression of the intermediate filament including GFAP, vimentin and nestin in Mȕller cells [18]. In order to examine whether fenofibrate could affect gliosis in Mȕller cells, retinas were immunostained with GFAP. The results demonstrated that GFAP was virtually limited to the ganglion cell layer (GCL) and absent in IPL on non-diabetic retinas as shown in Fig. 3a. However, in diabetic retinas, increased GFAP immunoreactivity was observed in cell endfeet in the GCL and cell processes in the IPL of Mȕller cells (Fig. 3a). In addition, it is worthy to note that Nrf2 staining was also partially colocalized with GFAP-positive processes in IPL, suggesting that Nrf2 was expressed by activated Mȕller cells in diabetic retinas. Notably, fenofibrate treatment accentuated expression of Nrf2 and reversed abnormal pattern of GFAP in diabetic retinas (Fig. 3a). Furthermore, GFAP expression was also assessed by Western blot analysis. As shown in Fig. 3b, diabetic mice exhibited increased retinal GFAP expression as compared with non-diabetic controls. Administration of fenofibrate by daily diet abolished diabetes-upregulated GFAP expression in retinas (Fig. 3b). Taken together, these findings demonstrated that activation of Mȕller cell gliosis is involved in DR and fenofibrate may protect retinas against diabetes-induced detrimental Mȕller gliosis.

Upregulation of Nrf2 by fenofibrate inhibits glial activation in diabetic retinas. Retinal expression and distribution of GFAP were determined by immunofluorescence (a) and western blot analysis (b), respectively. a Immunosignal of GFAP, a well-known marker of Muller glial reactivity, is restricted mainly along the retinal surface in the non-diabetic mice. GFAP expression was moderately increased in diabetic retinas and notably in the Mȕller cell processes extended from the inner retina into the outer retinal layers in the diabetic mice. Fenofibrate reversed diabetes-elicited abnormal distribution of GFAP. Magnification: ×400 and scale bar 50 μm. b Western blot analysis showed that GFAP expression was significantly increased in retinas of the diabetic mice compared with those of the non-diabetic controls, which were diminished by fenofibrate intervention. n = 4 for each group. **p < 0.01 versus Non-DM and ‡ p < 0.01 versus DM
Fenofibrates blocked retinal NLRP3 inflammasome activation
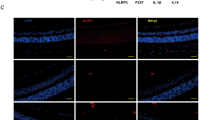
Inflammation is a characteristic feature of DR. Previous study indicated that NLRP3 inflammasome could be activated in a ROS-dependent manner, which links cellular oxidative stress to inflammatory response [10]. We next determined that whether fenofibrate could influence activation of NLRP3 inflammasome in diabetic retinas. Retinal NLPR3 expression was detected by Western blot analysis and semi-quantified by densitometry. As shown in Fig. 4a, NLRP3 expression was increased in retinas of diabetic mice; however, a significant reduction of NLRP3 was observed in retinas of fenofibrate-treated diabetic mice compared to vehicle-treated ones. NLRP3 inflammasome could recruit and trigger auto-proteolytic cleavage of pro-caspase-1 to generate active caspase-1. As shown in Fig. 4b, the active caspase-1 p20 subunit was substantially increased in the diabetic retinas, which was abolished by fenofibrate treatment. Moreover, double-labeling experiments revealed that retinal caspase-1 was mainly colocalized with Mȕller cell inner process and markedly increased by diabetes (Fig. 4c), suggesting a central role of Mȕller cell-derived inflammatory response in diabetic retinas. In addition, active caspase-1 is essential for the cleavage and maturation of pro-interleukin 1β (Pro-IL-1β). As shown in Fig. 4d, the mature IL-1β p17 form was markedly increased in the diabetic retinas. Fenofibrate treatment significantly attenuated up-regulation of active IL-1β p17 form in the diabetic retinas (Fig. 4d). These data suggested that fenofibrate may exert its anti-inflammatory effect on DR via inhibiting activation of NLRP3 inflammasome.

Fenofibrate suppressed retinal inflammasome formation. Retinal inflammasome formation was determined by expression of NLRP3, active Caspase-1 p20 and IL-1β p17. a Retinal NLRP3 levels were increased in the diabetic mice compared with the non-diabetic control, which was reversed by fenofibrate treatment. b Caspase-1 p20 was dramatically increased in the diabetic mouse retinas, which was attenuated by fenofibrate treatment. c Retinal distribution of Caspase-1 was detected by immunostaining. Weak expression of Caspase-1 was observed in the non-diabetic retinas. Of note, Caspase-1 was markedly increased in diabetic retinas and mainly colocalized with GS. Fenofibrate diminished diabetic-induced Caspase-1 immunoreactivity. Magnification: ×400 and scale bar 50 μm. d Retinal IL-1β p17 was strikingly increased in the diabetic mouse retinas, which was significantly reduced in the fenofibrate-treated diabetic mice compared with the vehicle-treated ones. n = 4. **p < 0.01 versus Non-DM and † p < 0.05 or ‡ p < 0.01 versus DM
Fenofibrate ameliorated retinal leukostasis and vascular leakage in diabetic mice
IL-1β is a potent pro-inflammatory cytokine, which stimulates expression of adhesion molecules such as ICAM-1 in vascular endothelial cells. As expected, diabetic retinas showed a higher level of ICAM-1 (Fig. 5a). Upregulated ICAM-1 could mediate leukocyte adherence to the retinal vasculature. We next performed Concanavalin A lectin staining to visualize the retinal leukostasis. The result demonstrated that there were more leukocytes adherent to retinal vasculature of diabetic mice compared with non-diabetic controls (Fig. 5b). Fenofibrate significantly decreased retinal ICAM-1 expression and reduced adherent leukocytes in retinal vessels in the diabetic mice (Fig. 5a, b). Furthermore, adherent leukocytes may compromise vascular endothelial integrity and result in retinal vascular leakage. Then we quantified retinal vascular permeability by Evan’s blue albumin method. Evan’s blue dye has high affinity with albumin and been widely used for quantification of albumin leakage across the blood–retinal barrier. As shown in Fig. 5c, a marked increase of extravasation of the albumin-bond Evan’s blue was observed in diabetic retinas, indicating that diabetes broke the blood–retinal barrier and led to retinal vascular hyper-permeability. However, fenofibrate treatment significantly lowered retinal content of albumin-bond Evan’s blue in diabetic mice (Fig. 5c), which suggested that fenofibrate may protect retinas from diabetes-induced vascular leakage. Taken together, fenofibrate demonstrated a beneficial effect on diabetic vasculopathy.

Fenofibrate ameliorated vasculopathy in diabetic mice. a Retinal expression of ICAM-1 was determined by western blot analysis and semi-quantified by densitometry. Retinal ICAM-1 expression was significantly increased in the diabetic mouse retinas. Fenofibrate markedly inhibited diabetes-induced ICAM-1 expression in the retinas. b Diabetes-associated inflammation mediated circulating leukocytes (yellow arrow) adherent to retinal vasculature. Fenofibrate dramatically reduced the number of adherent leukocytes in diabetic mouse retinas. c Evan’s blue-albumin leakage was significantly increased in the retina of diabetic mice. Fenofibrate treatment significantly ameliorated Evan’s Blue extravasation in the retinas of diabetic mice. n = 4. **p < 0.01 versus Non-DM and † p < 0.05 or ‡ p < 0.01 versus DM
Discussion
Oxidative stress and inflammation are two major hallmarks of DR. In this study, we explored detailed mechanisms of fenofibrate’s anti-oxidative and anti-inflammatory effects on DR. Previous study revealed that photoreceptors are the major source of ROS generation in the retinas, and genetic or pharmaceutical elimination of photoreceptors inhibit diabetes-induced production of superoxide and inflammatory cytokines in retinas [19]. In parallel, we observed that inner and outer segments of photoreceptors generated highest levels of ROS in diabetic retinas. Inner segments (IS) and outer segments (OS) lie between photoreceptor cell nuclei and RPE microvilli. OS contain disk membranes responsible for visual cycling. IS provide the metabolic support for the OS. The proximal region of the IS, known as the myoid zone, contains plenty of Golgi apparatus for protein synthesis, whereas the distal region of the IS, known as ellipsoid zone, contains large amounts of mitochondria for energy production, indicating that photoreceptors have high metabolic demands during visual perception. Diabetes could compromise Mitochondrial function to trigger excessive ROS generation in retinas. CellROX is a fluorescent ROS-detection probe. In the present study, we found that CellROX fluorescence was weakly detected in non-diabetic retinas. However, in diabetic retinas, CellROX may be oxidized and bound with Mitochondrial DNA of the ellipsoid zone to exhibit bright fluorescence at IS and OS of retinal photoreceptors. Notably, fenofibrate inhibited ROS generation in the retina with diabetes. These findings provide us evidence that the protective effect of fenofibrate on diabetic retinas may attribute to its ant anti-oxidative capacity. Diabetic Retinopathy Study (DRS) established pan-retinal photocoagulation (PRP) as the standard treatment for PDR [20]. The Early Treatment for Diabetic Retinopathy Study (ETDRS) also showed that focal or grid photocoagulation applied to the macular reduced risk of moderate vision loss from clinically significant diabetic macular edema and scattered or pan-retinal photocoagulation applied to peripheral retinas reduced risk of severe vision loss from PDR [21]. Retinal laser coagulation destroys energy-consuming photoreceptors and reduces the oxygen consumption in the outer retina, thereafter attenuates hypoxia of inner retinas, blocks demanding signals for pro-angiogenic factor production, and causes regression of retinal new vessels. FIELD study demonstrated that there was a 32% reduction in the rate of laser photocoagulation for DR with fenofibrate treatment [1]. Based on our study, fenofibrate inhibiting photoreceptor ROS generation may provide the reasonable explanation for lower rate of laser requirement in diabetic patients with fenofibrate treatment.
Nrf2 signaling has been demonstrated to be activated in the stressed retinas [22, 23]. Deficiency of Nrf2 displayed early onset retinal vascular leakage and exacerbated neuronal dysfunction in diabetes [24]. In contrast, activation of Nrf2 protected retinas from oxidative damage [25, 26]. Previous study reported that fenofibrate activated Nrf2 by inducing Keap-1 degradation through an autophagy-dependent manner [27]. In this study, we demonstrated that fenofibrate markedly enhanced retinal Mȕller Nrf2 expression in diabetes. Meanwhile, expression of Nrf2 target gene including NQO-1 and HO-1 was also further increased by fenofibrate intervention. All these proved that fenofibrate may promote Nrf2 activation to combat oxidative stress in diabetic retinas. Several elegant studies demonstrated that PPARα was responsible for fenofibrate’s protective effect on diabetic retinopathy [28, 29]. However, whether PPARa is involved in Nrf2 signaling during DR needs further studies to explore.
Mȕller cells, the specialized glial cells in retinas, spread through the whole retina thickness from the inner to the outer limiting membrane. Mȕller cells contact with retinal neuron and vessels. Thus, they support retinal neurons through releasing neurotrophic factors, providing anti-oxidative support, recycling neurotransmitters and ions, and are also involved in regulation of blood retinal barrier [30]. Distinct from ubiquitous expression of GS by the Mȕller cells, GFAP was not constitutively expressed in quiescent Mȕller cells. Inducible GFAP expression could be elicited in Mȕller cells by tissue stress in a process termed of reactive gliosis. Mȕller glial activation or reactive gliosis could be detrimental and resulted in neuron damage and microvascular dysfunction during diabetes [31, 32]. In our study, diabetic mice developed mild to moderate reactive gliosis at age of 8 months. These findings provided indirect evidence that long-term diabetes may compromise retinal glia cells, which may be implicated in diabetic retinal neuropathy. Fenofibrate intervention abolished diabetes-induced Mȕller cells gliosis. Therefore, this study still highlights a protective role of fenofibrate on diabetic retinal neuropathy, which is worthy of future clinical trial to verify.
NLRP3 inflammasome could sense a wide range of endogenous metabolic disturbance to induce sterile inflammation [33, 34]. Upon activation, NLRP3 inflammasome forms a multi-protein complex that triggers the self-cleavage of caspase-1 and subsequently maturation and secretion of pro-inflammatory cytokines IL-1β. Inhibition of NLRP3 inflammasome was reported to ameliorate DR [35, 36]. In our study, we investigated expression and localization of caspase-1 in retinas and revealed that retinal caspase-1 was mainly expressed by Mȕller glias and activated by diabetes. The finding indicated that IL-β is mainly released by retinal Mȕller cells during diabetes, which is consistent with the previous finding that retinal Mȕller glial cells are a major source of pro-inflammatory cytokine production [37, 38]. Mature IL-1β may bind with its receptor on vascular endothelial cells in a paracrine manner to upregulate ICAM-1 express on the side of vascular lumen. ICAM-1-mediated adhesion of circulating leukocytes to retinal vasculature resulted in or even exaggerated vascular damage. In this study, we reported that fenofibrate inhibited NLRP3 inflammasome activation and thus attenuated vascular damage in DR and this finding enriched mechanisms for fenofibrate’s anti-inflammatory activity. Meanwhile, our finding emphasized a central role of Mȕller glial cells in maintaining homeostasis of “neurovascular unit” during DR.
Conclusion
Our data indicated that oxidative stress and inflammasome activation plays a pathogenic role in DR. Activation of Nrf2 signaling in Mȕller cells could be considered as a compensatory response to combat oxidative stress and detrimental reactive gliosis during diabetic retinopathy. Fenofibrate could attenuate oxidative stress in diabetic retinas via further activating Nrf2 signaling and upregulating downstream anti-oxidative gene expression. In addition, fenofibrate also exert its protective effect on diabetic retinas via inhibiting NLRP3 inflammasome activation.
References
Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d’Emden MC, Crimet DC, O’Connell RL, Colman PG, Investigators FS (2007) Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 370:1687–1697. https://doi.org/10.1016/S0140-6736(07)61607-9
Group AS, Group AES, Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, Hubbard L, Esser BA, Lovato JF, Perdue LH, Goff DC Jr., Cushman WC, Ginsberg HN, Elam MB, Genuth S, Gerstein HC, Schubart U, Fine LJ (2010) Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 363:233–244. https://doi.org/10.1056/NEJMoa1001288
Noonan JE, Jenkins AJ, Ma JX, Keech AC, Wang JJ, Lamoureux EL (2013) An update on the molecular actions of fenofibrate and its clinical effects on diabetic retinopathy and other microvascular end points in patients with diabetes. Diabetes 62:3968–3975. https://doi.org/10.2337/db13-0800
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Cheng Y, Zhang J, Guo W, Li F, Sun W, Chen J, Zhang C, Lu X, Tan Y, Feng W, Fu Y, Liu GC, Xu Z, Cai L (2016) Up-regulation of Nrf2 is involved in FGF21-mediated fenofibrate protection against type 1 diabetic nephropathy. Free Radic Biol Med 93:94–109. https://doi.org/10.1016/j.freeradbiomed.2016.02.002
Ibarra-Lara L, Hong E, Soria-Castro E, Torres-Narvaez JC, Perez-Severiano F, Del Valle-Mondragon L, Cervantes-Perez LG, Ramirez-Ortega M, Pastelin-Hernandez GS, Sanchez-Mendoza A (2012) Clofibrate PPARalpha activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J Cardiovasc Pharmacol 60:323–334. https://doi.org/10.1097/FJC.0b013e31826216ed
Ruiz S, Pergola PE, Zager RA, Vaziri ND (2013) Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int 83:1029–1041. https://doi.org/10.1038/ki.2012.439
Chen WJ, Wu C, Xu Z, Kuse Y, Hara H, Duh EJ (2017) Nrf2 protects photoreceptor cells from photo-oxidative stress induced by blue light. Exp Eye Res 154:151–158. https://doi.org/10.1016/j.exer.2016.12.001
Ogura Y, Sutterwala FS, Flavell RA (2006) The inflammasome: first line of the immune response to cell stress. Cell 126:659–662. https://doi.org/10.1016/j.cell.2006.08.002
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–140. https://doi.org/10.1038/ni.1831
Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A (2017) NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. https://doi.org/10.1111/aos.13427
Deng Y, Han X, Yao Z, Sun Y, Yu J, Cai J, Ren G, Jiang G, Han F (2017) PPARalpha agonist stimulated angiogenesis by improving endothelial precursor cell function via a NLRP3 inflammasome pathway. Cell Physiol Biochem 42:2255–2266. https://doi.org/10.1159/000479999
Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX (2012) Activation of endoplasmic reticulum stress by hyperglycemia is essential for Muller cell-derived inflammatory cytokine production in diabetes. Diabetes 61:492–504. https://doi.org/10.2337/db11-0315
Cheng R, Ding L, He X, Takahashi Y, Ma JX (2016) Interaction of PPARalpha with the canonic Wnt pathway in the regulation of renal fibrosis. Diabetes 65:3730–3743. https://doi.org/10.2337/db16-0426
Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX (2010) Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes 59:1528–1538. https://doi.org/10.2337/db09-1057
Akiyama G, Azuchi Y, Guo X, Noro T, Kimura A, Harada C, Namekata K, Harada T (2017) Edaravone prevents retinal degeneration in adult mice following optic nerve injury. Invest Ophthalmol Vis Sci 58:4908–4914. https://doi.org/10.1167/iovs.17-22250
Li J, Wang JJ, Zhang SX (2011) Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J Biol Chem 286:4912–4921. https://doi.org/10.1074/jbc.M110.199729
Lewis GP, Fisher SK (2003) Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int Rev Cytol 230:263–290
Du Y, Veenstra A, Palczewski K, Kern TS (2013) Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci USA 110:16586–16591. https://doi.org/10.1073/pnas.1314575110
No Authors (1987) Indications for photocoagulation treatment of diabetic retinopathy: Diabetic Retinopathy Study Report no. 14. The Diabetic Retinopathy Study Research Group. Int Ophthalmol Clin 27:239–253
No Authors (1991) Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 98:766–785
Wei Y, Gong J, Xu Z, Duh EJ (2016) Nrf2 promotes reparative angiogenesis through regulation of NADPH oxidase-2 in oxygen-induced retinopathy. Free Radic Biol Med 99:234–243. https://doi.org/10.1016/j.freeradbiomed.2016.08.013
Wei Y, Gong J, Xu Z, Thimmulappa RK, Mitchell KL, Welsbie DS, Biswal S, Duh EJ (2015) Nrf2 in ischemic neurons promotes retinal vascular regeneration through regulation of semaphorin 6A. Proc Natl Acad Sci USA 112:E6927–E6936. https://doi.org/10.1073/pnas.1512683112
Xu Z, Wei Y, Gong J, Cho H, Park JK, Sung ER, Huang H, Wu L, Eberhart C, Handa JT, Du Y, Kern TS, Thimmulappa R, Barber AJ, Biswal S, Duh EJ (2014) NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 57:204–213. https://doi.org/10.1007/s00125-013-3093-8
Inoue Y, Shimazawa M, Noda Y, Nagano R, Otsuka T, Kuse Y, Nakano Y, Tsuruma K, Nakagami Y, Hara H (2017) RS9, a novel Nrf2 activator, attenuates light-induced death of cells of photoreceptor cells and Muller glia cells. J Neurochem 141:750–765. https://doi.org/10.1111/jnc.14029
Sun Y, Xiu C, Liu W, Tao Y, Wang J, Qu YI (2016) Grape seed proanthocyanidin extract protects the retina against early diabetic injury by activating the Nrf2 pathway. Exp Ther Med 11:1253–1258. https://doi.org/10.3892/etm.2016.3033
Park JS, Kang DH, Lee DH, Bae SH (2015) Fenofibrate activates Nrf2 through p62-dependent Keap1 degradation. Biochem Biophys Res Commun 465:542–547. https://doi.org/10.1016/j.bbrc.2015.08.056
Ding L, Cheng R, Hu Y, Takahashi Y, Jenkins AJ, Keech AC, Humphries KM, Gu X, Elliott MH, Xia X, Ma JX (2014) Peroxisome proliferator-activated receptor alpha protects capillary pericytes in the retina. Am J Pathol 184:2709–2720. https://doi.org/10.1016/j.ajpath.2014.06.021
Chen Y, Hu Y, Lin M, Jenkins AJ, Keech AC, Mott R, Lyons TJ, Ma JX (2013) Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 62:261–272. https://doi.org/10.2337/db11-0413
Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A (2006) Muller cells in the healthy and diseased retina. Prog Retin Eye Res 25:397–424. https://doi.org/10.1016/j.preteyeres.2006.05.003
Sorrentino FS, Allkabes M, Salsini G, Bonifazzi C, Perri P (2016) The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci 162:54–59. https://doi.org/10.1016/j.lfs.2016.08.001
Moran EP, Wang Z, Chen J, Sapieha P, Smith LE, Ma JX (2016) Neurovascular cross talk in diabetic retinopathy: pathophysiological roles and therapeutic implications. Am J Physiol Heart Circ Physiol 311:H738–H749. https://doi.org/10.1152/ajpheart.00005.2016
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867. https://doi.org/10.1038/nature05485
Rampanelli E, Orso E, Ochodnicky P, Liebisch G, Bakker PJ, Claessen N, Butter LM, van den Bergh Weerman MA, Florquin S, Schmitz G, Leemans JC (2017) Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation. Sci Rep 7:2861. https://doi.org/10.1038/s41598-017-01994-9
Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, Lu L, Xu X, Guan H, Zheng Z, Qiu Q (2017) Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res 66:157–166. https://doi.org/10.1007/s00011-016-1002-6
Yin Y, Chen F, Wang W, Wang H, Zhang X (2017) Resolvin D1 inhibits inflammatory response in STZ-induced diabetic retinopathy rats: Possible involvement of NLRP3 inflammasome and NF-kappaB signaling pathway. Mol Vis 23:242–250
Eastlake K, Banerjee PJ, Angbohang A, Charteris DG, Khaw PT, Limb GA (2016) Muller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 64:495–506. https://doi.org/10.1002/glia.22942
Le YZ (2017) VEGF production and signaling in Muller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vis Res. https://doi.org/10.1016/j.visres.2017.05.005
Funding
This study was supported by NSFC Grants 81400427, 81460163, 81741058 and 81300786; Young Talent Scholar Grant 2016KJXX-12; FRFCU Grant xjj2015015; RFDP grant 20133601120012; Research Grants from Jiangxi Education Department GJJ14094, GJJ13175; Research Grants from Jiangxi Science and Technology Department 20142BDH80005, 20142BAB215029, 20132BAB205024. Parts of this study were orally presented at annual meeting of Asia-Pacific Academy of Ophthalmology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interests to declare.
Additional information
Qiuping Liu and Fengjun Zhang have contributed equally to this study.
Rights and permissions
About this article

Cite this article
Liu, Q., Zhang, F., Zhang, X. et al. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem 445, 105–115 (2018). https://doi.org/10.1007/s11010-017-3256-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-017-3256-x