
Abstract
The Wnt signaling pathway plays a predominant role in aberrant proliferation in myriad of cancers. In non-cancerous cells, Wnts are blocked by the secreted frizzled-related proteins (sFRPs) that are generally downregulated in cancer cells. We have purified and characterized bacterially expressed glutathione S-transferase-tagged SFRP4 from a novel clone generated from human cell origin. Cervical cancer (HeLa) and lung cancer (A549) cells, in which Wnt and associated genes were found to be expressed, were treated with the purified recombinant sFRP4, which revealed a significant dose-dependent cell growth inhibition up to 40 %. The current investigation on functionality of this bacterially produced recombinant sFRP4 in arresting cancer cell proliferation is the first of its kind, where G2/M phase arrest and early apoptosis were evident. Increase in phosphorylated β-catenin in sFRP4 treatment indicated inhibition of Wnt pathway, which was further confirmed by downregulation of pro-proliferative genes, namely cyclin D1, c-myc, and survivin. Functional activity of recombinant sFRP4 was further exploited in co-therapy module with chemotherapeutic drugs to decipher molecular events. Collectively, our study on purified recombinant sFRP4 from bacterial host holds great promise in targeting Wnt signaling for exploring new strategies to combat cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aberrant Wnt signaling causes increased proliferation and reduced apoptosis in a multitude of cancers. Understanding the underlying details of this pathway may prove to be pivotal for the future of cancer therapy [1]. This may involve binding of inhibitory molecules to Wnt ligands to prevent its functioning via canonical and non-canonical pathways [2, 3] or targeting downstream entities in the canonical Wnt pathway [4]. One such evolving prospect in its very nascent stage of development is the family of secreted antagonists of Wnt signaling, called the secreted frizzled-related proteins (sFRPs). sFRP4 is one of its prominent isoforms, having serious implications in tumorigenesis [5]. In normal adult cells, it binds to Wnt morphogens and prevents their binding to corresponding transmembrane Frizzled receptor, thereby impeding the canonical Wnt pathway. However, loss of expression of sFRP4 due to promoter hypermethylation has been found in several types of cancer, such as colorectal cancer [6], bladder cancer [7], mesothelioma [8], cervical cancer [9], and ovarian cancer [10]. This promotes the upregulation of Wnt signaling, resulting in stabilization of a cytoplasmic pool of β-catenin protein, which is the signaling node of the canonical Wnt pathway. β-Catenin, in turn, transcriptionally activates pro-proliferative genes like cyclin D1 [11], survivin [12], and c-myc [13], thereby deregulating cell proliferation. In non-cancerous cells, sFRPs block the Wnts; subsequently, β-catenin is phosphorylated and marked for degradation, leading to downregulation of pro-proliferative genes. This gives rise to the possibility of mimicking this scenario in case of cancer cells as well by blocking the Wnt ligands either with functional recombinant sFRP4 added to the cell culture media. If such a system is designed, alterations in expression levels of downstream molecules may be used as a tool for quantitative estimation of the extent of inhibition of the Wnt pathway. Moreover, it raises the question of clinical application of the recombinant sFRP4; if aberrant proliferation of cancer cells can be restricted by blocking the Wnt pathway with sFRP4, then this protein may have a great potential in the field of recombinant protein therapeutics.
Recent investigations into interactions of sFRP4 with several Wnt ligands have created a surge of interest in exploring its anti-proliferative activity. Both transfection studies and exogenous addition of sFRP4 to culture have demonstrated that it is accountable for keratinocyte differentiation and apoptosis [14]. In breast cancer cell line MCF-7, sFRP4-conditioned media were found to diminish cell proliferation and downregulated Wnt signaling genes [15]. In a similar study, transfection of sFRP4 was found to reduce proliferation via the canonical Wnt pathway in prostate cancer cells [16]. Interestingly, it has also been shown to induce apoptosis in β-catenin-deficient mesothelioma cells, suggesting that canonical Wnt pathway-independent signaling phenomena may also be at play [8]. However, intriguing discrepancies have been reported, challenging the generally accepted antagonistic function of sFRPs [17]. For instance, sFRP4 was found overexpressed in colorectal cancer patient samples, revealing that sFRP4 may have entirely different biological roles in different cancer types [18]. These investigations reflect the bewilderingly complex behavior of this signaling system. The fact that it varies so widely in different cell types and growth conditions necessitates further explorations into this field.
Another compelling facet of sFRP4 that has surfaced is its ability to chemosensitize cancer cells to conventional drugs. So far documented in ovarian cancer cells [19] and glioma stem cells [20], this phenomenon has the potential to find a place for sFRP4 in the transforming field of combination therapy, which could reduce the exacerbated side effects of chemotherapeutic agents, while enhancing the anticancer efficacy. However, the same may not be applicable for bacterially expressed recombinant sFRP4 or in case of other forms of cancer. If the Wnt blocking efficiency of bacterially produced sFRP4 is retained in conjunction with drugs, while the drugs induce apoptosis via other interconnected signaling pathways, then this system may indeed prove to be successful in a combination module.
From the structure point of view, the N-terminal cysteine-rich domain (CRD) is responsible for binding to Wnt. Each of the sFRPs, including sFRP4, has the CRD, which shares sequence similarity with CRD of Frizzled receptors, thus serving as putative binding sites for Wnt ligands [17]. The C-terminal domain, called the netrin-like domain (NLD), may also function in promoting cell death, which may or may not be via canonical Wnt pathway [21, 22]. While not much is known about the NLD, functional CRD alone has been shown to be sufficient to suppress Wnt signaling [23]. Posttranslational modifications in the form of N-linked glycosylations have been found in the sFRPs, a number of putative sites being in the NLD [24]. Although a few potential sites are present in the CRD as well, glycosylation is not essential for binding to Wnt ligand [25]. This suggested that human sFRP4 expressed in bacterial system may retain its functionality. If the limitation of eukaryotic protein folding can be overcome, expression in bacterial system has the advantage of lower cost and potential for production in bulk, which makes it a lucrative option for potential therapeutic applications.
In this investigation, we have cloned the sFRP4 gene from a novel source into bacterial expression vector pGEX-4T2 containing glutathione S-transferase (GST) tag. The GST-sFRP4 protein, purified from Escherichia coli BL21 (DE3) cells, was characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI TOF/TOF), circular dichroism spectra, and Western blot analyses. We assessed the functionality of recombinant protein on two different cancer cell lines—cervical carcinoma (HeLa) and lung carcinoma (A549). To the best of our knowledge, this is the first report interpreting the structural and functional characterization of bacterially expressed GST-sFRP4. Further, targeting of canonical Wnt pathway was proven by analyzing levels of β-catenin and phosphorylated β-catenin protein as well as gene expression analysis of cyclin D1, survivin, and c-myc. In addition, combination module was designed with traditional chemotherapeutic drugs for augmented efficiency, which also showed that functionality of sFRP4 in blocking the Wnt pathway was retained even in conjunction with drugs. Anticancer effect was ascertained by cell viability assay-, dual staining-, and flow cytometry-based cell cycle and apoptosis analyses. These data inspire the possibility of exploiting this recombinant sFRP4 in the avenue of possible protein therapeutics.
Materials and methods
Cell culture
Cervical cancer (HeLa), lung cancer (A549), renal cancer (ACHN), and non-cancerous human embryonal kidney (HEK-293) cells were purchased from National Centre for Cell Science, India. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), with 10 % FBS, 100 U/ml penicillin, 10 mg/ml streptomycin, in 5 % carbon dioxide incubator with controlled humidity at 37 °C.
Materials
All items were purchased from Sigma Aldrich unless mentioned otherwise.
Cloning of sFRP4
Total RNA was isolated from human renal cell carcinoma cell line (ACHN) using Tri reagent. cDNA was synthesized with Verso cDNA Kit (Thermo Scientific), according to manufacturer’s protocol. The 1041-bp gene of sFRP4 was amplified from the cDNA pool using forward primer 5′-ATGTTCCTCTCCATCCTAGTGGCGCT-3′ and reverse primer 5′-GCTCACACTCTTTTCGGGTTTGTTCTC-3′. The gene was incorporated into pGEMT-Easy vector (Promega) after overnight ligation at 4 °C. From this clone, sFRP4 gene was PCR-amplified with gene-specific forward primer 5′-GGCGGATCCATGTTCCTCTCCATCCT-3′ and reverse primer 5′-GCCCTCGAGTCACACTCTTTTCGGGT-3′ containing overhangs for BamHI and XhoI restriction sites. Subsequently, the amplified product was subcloned into expression vector pGEX-4T2 by digestion with BamHI and XhoI restriction enzymes, followed by its ligation of digested vector at 37 °C. Ligated product was transformed into E. coli DH5α. Clone was confirmed by sequencing at Xcelris Labs Ltd., India.
Expression of sFRP4 in E. coli BL21(DE3)
Clone was transformed into E. coli BL21(DE3), which is suitable for protein expression. Primary culture was grown in Luria–Bertani media overnight at 37 °C under shaking conditions. Secondary culture was given with 1 % of primary culture as inoculum. When an OD of 0.6 was attained, sFRP4 protein was induced by IPTG (isopropyl β-d-1-thiogalactopyranoside). Expression was optimized by varying induction time, temperature, shaking speed, and concentration of IPTG. Maximum expression was obtained at induction temperature and time of 28 °C and 8 h, respectively. However, upon lysis of the bacterial cells with 10 mM phosphate-buffered saline (PBS), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM ethylenediaminetetraacetic acid (EDTA), protein was found to be expressed entirely as inclusion bodies.
Purification of sFRP4 from bacterial system
For the purpose of solubilizing sFRP4 protein from the inclusion bodies, our initial attempts involved varying induction time, temperature, and concentration of IPTG. When we found that the protein was still expressed as insoluble fraction, we transformed the pGEX-4T2 holding sFRP4 gene into different strains of bacteria, viz Rosetta-gami and E. coli BL21 pLysS. When we still did not get the desired results, we modified the composition of the lysis buffer by using various detergents, such as Triton X-100, sodium deoxycholate, and N-lauroylsarcosine. Eventually, a concentration of 0.32 % N-lauroylsarcosine added to the lysis buffer composition yielded in the entire protein being expressed in the soluble fraction. To optimize binding of GST-sFRP4 to the glutathione-agarose affinity chromatography column, the supernatant obtained after centrifugation of lysed cells was stirred in cold for 1 h, after addition of 1 % Triton X-100. This was then diluted two times with 10 mM PBS, filtered, and loaded into the column in three batches, with 30 min incubation time each. Subsequently, the column was washed 12 times with PBS, at the end of which sFRP4 was eluted with 5 mL elution buffer (50 mM Tris, pH 9.5, and 15 mM L-reduced glutathione). Elution fractions were electrophoresed in a 12 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to observe expression of GST-sFRP4 in the purified elute. Further, the induction temperature and time were reduced to 24 °C and 6 h, respectively, in order to prevent degradation of GST from GST-sFRP4. Before performing each of the experiments, protein was dialyzed in stepwise manner against Tris–HCl buffer (pH 7.4), where the final buffer concentration was maintained as 10 mM. Bradford assay was performed to estimate the protein concentration, using bovine serum albumin as standard.
Homology modeling and docking analyses
The structure of the binding domain (CRD or cysteine-rich domain) of sFRP4 was predicted using PHYRE2 Protein Fold Recognition Server, along with those of sFRP4 and GST-sFRP4 [26]. Another protein structure prediction server I-TASSER [27–29] was used to predict the 3D structure of WNT7A, which has been documented to bind to sFRP4 [30]. To ascertain whether the CRD remained intact in case of sFRP4 and GST-sFRP4 structures, CRD was individually overlapped with each of the two, using the molecular visualization system PyMOL, and the root mean square deviation value was generated in each case. Also, CRD of sFRP4 was aligned with the crystal structure of sFRP3 (PDB ID: 1ijxA), to verify the integrity of the modeled structure. Thereafter, WNT7A was docked with each of the three modeled structures, namely CRD-SFRP4, sFRP4, and GST-sFRP4, using ClusPro server [31–34]. The docked structures were analyzed with PDBsum Generate [35, 36].
MALDI TOF/TOF analysis
Confirmation of sFRP4 sequence was done by MALDI TOF/TOF analysis. In situ gel tryptic digestion of GST-sFRP4 was performed using Trypsin Profile In-Gel Digestion Kit (Sigma) following manufacturer’s protocol. Sample was mixed with 10 mg/ml α-cyano-4-hydroxycinnamic acid matrix in 0.1 % trifluoroacetic acid, 50 % acetonitrile, and spotted on the MALDI target plate. Eventually, it was analyzed with 4700 Proteomics Analyzer with TOF/TOF Optics (Applied Biosystems) equipped with a diode-pumped solid-state class I laser and MS/MS data were acquired in automatic mode.
Secondary structure analysis using circular dichroism
Formation of secondary structure of GST-sFRP4 was confirmed by circular dichroism analysis using a JASCO-815 spectrometer (Jasco, Japan). Purified and dialyzed protein was analyzed in a cuvette of 1-mm path length, under constant flow of nitrogen gas at a rate of 5 L/min and maintenance of constant temperature at 25 °C. Sample was scanned at a speed of 50 nm/min, from wavelength 250 nm to 180 nm. Background subtraction was done with Tris buffer of concentration equal to that of the protein sample.
Expression profile analysis of genes involved in the Wnt pathway in three different cell lines
The expression levels of six genes relevant to the Wnt signaling cascade were checked in three different cancer cell lines, namely HeLa, A549, and one non-cancerous cell line HEK-293. The genes analyzed were Wnt4, Wnt7b, Wnt10b, co-receptor LRP6, and β-catenin. Based on the results obtained in this expression profiling study, we proceeded with our work.
Western blotting
The technique of Western blotting was applied to assess whether the purified protein was indeed GST-sFRP4 using anti-GST antibody. Purified GST-sFRP4 was electrophoresed on 12 % SDS-PAGE. Also, the effect of GST-sFRP4 on the Wnt pathway of cancer cells was probed using antibodies against β-catenin as well as its phosphorylated form. In this pursuit, cells were treated with GST-sFRP4 for 24 h. Thereafter, whole cell lysate was prepared from treated and untreated cells, with RIPA buffer, supplemented with 1 mM PMSF and 1 mM EDTA. Total protein content was estimated with Lowry assay, and 12 % SDS-PAGE was done after loading equal amounts of protein for each sample. In all cases, protein was electroblotted onto a PVDF membrane for 3 h at constant voltage, followed by blocking with 4 % BSA in PBST for 2 h. Next, the membrane was incubated overnight with the respective primary antibodies under cold conditions. Subsequently, membrane was washed six times with PBST, before being incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h. Then, it was washed six times with PBST and developed with chemiluminescence peroxidase substrate kit (Sigma). It should be mentioned here that for probing phospho-β-catenin, TBST was used in all steps, instead of PBST.
Combination therapy
Combination therapy of GST-sFRP4 with conventional chemotherapeutic agents, cisplatin and doxorubicin, was attempted. For all of the following experiments, this mode of co-therapy was employed.
Cell viability assay
Cell viability assay was conducted in order to assess the effect of GST-sFRP4 and the co-therapy regime on HeLa and A549 cells. For this purpose, cells were seeded at a density of 7000 cells per well in 96-well plate and allowed to attach for about 8 h. Subsequently, serum media was removed, and treatment was done with the dialyzed and quantified GST-sFRP4 (0 nM to 32 nM for HeLa and 0 to 20 nM for A549), alone and in combination with cisplatin (0 µM to 16 µM) or doxorubicin (0 µM to 0.6 µM) in serum-free media for 48 h. At the end of the treatment period, MTT assay was performed, whereby live cells converted MTT to purple formazan crystals, which were dissolved by adding DMSO, and absorbance was determined at 550 nm. Percentage of viable cells was calculated using the formula:
Control experiments were also performed using a Wnt pathway inhibitor XAV939 to show that the Wnt pathway is indeed hyperactive in HeLa and A549 cells.
Cell cycle analysis
Flow cytometric analysis of cell cycle was performed with Fluorescence Activated Cells Sorter (FACSCalibur, BD Biosciences, NJ, USA) to ascertain the occurrence of cell cycle arrest, if any. Cells were seeded in six-well plates at a density of 105 cells per well. Treatment was given in the same manner as described previously. Concentrations of GST-sFRP4, cisplatin, and doxorubicin added to the cells were 12 nM, 7 µM, and 0.2 µM, respectively. After 48 h of treatment, cells were harvested by trypsinization and fixed with 70 % ethanol under chilled conditions. Cells were then washed with pre-chilled PBS, and RNaseA (Amresco) was added at a concentration of 0.2 mg/ml and incubated for 1 h at 37 °C. Next, cells were incubated with 10 µg/ml of the nucleic acid intercalating dye propidium iodide (PI) for 15 min in dark, before analysis.
Apoptosis detection assay
Apoptosis was detected by flow cytometry using Annexin V-fluorescein isothiocyanate (FITC)/PI Apoptosis Detection Kit (BD Biosciences). Cells were seeded and treated as mentioned previously. After the treatment period, cells were harvested, washed with PBS, and incubated with Annexin V-FITC and/or PI, following manufacturer’s protocol. Unstained cell samples were also kept for each mode of treatment to check auto-fluorescence. After staining for 30 min, cells were analyzed in BD FACSCalibur.
Acridine orange (AO)/ethidium bromide (EB) dual staining for detection of apoptosis
Dual staining of cells with AO and EB was done to distinguish between healthy and membrane compromised cells posttreatment. Cells were seeded in 96-well plate and treated with the concentrations of protein and/or drugs showing maximum cell-inhibitory effect in MTT assay. Then, media were discarded and cells were incubated with 2 µg/ml of AO and 10 µg/ml of EB in PBS for 5 min in the dark. Eventually, cells were washed with PBS before being visualized under a fluorescence microscope (Nikon ECLIPSE TS100).
Real-time PCR analysis
Real-time PCR analysis was performed to check the expression of crucial pro-proliferative genes downstream of the Wnt pathway, namely cyclin D1, survivin, and c-myc. In this regard, treatment of cells was done with same concentrations of protein/drugs as in the flow cytometry-based experiments. RNA was extracted from each sample with the Mammalian Total RNA Isolation Kit (Sigma), following manufacturer’s protocol. RNA was quantified with Nanodrop (GE Healthcare Life Sciences), and 1 µg equivalent cDNA was synthesized using First Strand cDNA Synthesis Kit (Thermo Scientific). With this cDNA as template, gene-specific primers (given in supporting information Table S1), and SYBR Green Mastermix, Real-time PCR (Rotor-Gene Q, Qaigen) was done.
Statistical tests
Statistical tests were performed using GraphPad Prism. Significance of data was determined by one-way or two-way ANOVA, as applicable.
Results
Expression profile analysis of genes associated with the Wnt family
The expression of six genes associated with the Wnt family was analyzed in three different cell lines (Fig. 1a). As the aim of this study was to target the aberrant Wnt signaling cascade with recombinant sFRP4, we wanted to be certain about the expression of Wnt ligands, β-catenin, and LRP6 in these cells. Out of the three Wnts, viz Wnt4, 7b, and 10b, at least two were found expressed in all cell lines screened; prominent expression of co-receptor LRP6, and β-catenin was also observed. Encouraged by these data, we proceeded to clone and purify sFRP4 in order to examine its role in blocking Wnt signaling for possible therapeutic applications.

a Expression profiling of genes belonging to Wnt signaling pathway by RT-PCR analysis of three different cell lines. MTT assay depicting anti-proliferative activity of Wnt/β-catenin pathway inhibitor XAV939 b on HeLa cells and c on A549 cells
In addition, a small molecule drug XAV939 (Fig. 1b, c) was employed, which solely inhibits the Wnt/β-catenin pathway [37]. Cell viability assays revealed that in both cell types, IC50 value was attained at approximately 100 μM of XAV939, confirming the functionality of Wnt pathway.
Cloning, expression, and purification of sFRP4
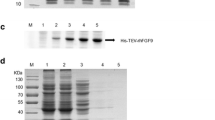
Cloning of sFRP4 was done from novel human source ACHN renal carcinoma cells following the procedure described in the “Materials and methods” section. As the expression level of sFRP4 in ACHN cells was low, re-PCR was done to obtain sufficient PCR product (Fig. 2a) for subsequent cloning steps. sFRP4 gene was cloned into the pGEX-4T2 via an intermediate TA vector pGEMT-Easy. Clone was confirmed in pGEX-4T2 by digestion with BamHI and XhoI restriction enzymes, which generated a band corresponding to 1041 bp of sFRP4 gene (Fig. 2b).

a Lane 1: DNA hyperladder, lane 2: sFRP4 amplified with gene-specific primers from cDNA pool of ACHN cell line. b Lane 1: DNA hyperladder, lane 2: uncut pGEX-4T2 containing sFRP4 gene, lane 3: release of band corresponding to 1041 bp of sFRP4 on digestion of sFRP4 pGEX-sFRP4 with BamHI and XhoI. c Expression of GST-sFRP4 in E. coli BL21(DE3), lane 1: protein marker, lane 2: uninduced pGEX-sFRP4, lane 3: supernatant after lysis of induced E. coli BL21 transformed with sFRP4 gene containing pGEX-4T2, lane 4: pellet obtained after lysis, d purification of GST-sFRP4, lane 1: protein marker, lane 2: band corresponding to 64 kDa of purified GST-sFRP4. e Tryptic digestion of GST-sFRP4 generated a peptide signature corresponding to the specific sequence of sFRP4, on MALDI TOF/TOF mass spectrometric analysis, f circular dichroism spectra depicting 20 % alpha helix and 11.9 % β-sheet
For expression of recombinant protein in bacterial system, we chose the common E. coli BL21 (DE3) strain. However, the recombinant protein was completely present as inclusion bodies. Therefore, we attempted to solubilize the protein by varying induction temperature, time, shaking speed, and concentration of IPTG. Strains specifically engineered for expression of mammalian proteins were also tried in order to solve this problem. Since none of the methods employed seemed to yield protein in soluble form, we proceeded to continue with BL21 (DE3) and solubilize the GST-sFRP4 using detergents [38, 39]. We observed optimum expression of GST-sFRP4 at 28 °C after carrying out induction at temperatures ranging from 16 to 37 °C. Varying the concentration of IPTG and induction time, optimum yield of protein was obtained for 0.5 mM IPTG and 6 h, respectively (Fig. 2c). To obtain the protein in soluble fraction, a number of strategies involving the usage of various detergents were implemented following the literature [38, 39]. Finally, 0.32 % of the ionic detergent sarkosyl was used to successfully solubilize the protein from inclusion bodies and mild stirring was done in 1 % Triton X-100. Triton X-100 is a nonionic detergent that probably sequestered the sarkosyl into its micelles and helped in renaturation of the protein that had been partially denatured by sarkosyl [40]. Eventual washing steps during purification and the subsequent process of dialysis also helped the removal of detergent from the system. Electrophoresis of the purified GST-sFRP4 yielded a single band at approximately 64 kDa (Fig. 2d). A faint band of GST was also observed at 26 kDa, which could be due to cleavage of the recombinant protein. Reduction of induction temperature from 28 to 24 °C was found to yield a fainter band of GST. Quantifying the purified GST-sFRP4 with Bradford assay demonstrated that the total yield of protein from 100 ml of culture was 0.3 mg. Stepwise dialysis against Tris–HCl buffer (pH 7.4) ensured gradual removal of detergents and renaturation of the protein.
Characterization of recombinant GST-sFRP4
Soft ionization techniques like MALDI coupled with TOF mass spectrometry have proven to be ideal tools for protein sequencing and fingerprinting. Here, enzymatic digestion of purified recombinant sFRP4 in situ yielded peptide fragments, which were analyzed in the MS/MS mode for confirmation of protein identity. The data file was analyzed with FindPept proteomics tool provided with ExPASy (http://web.expasy.org/findpept/) [41, 42], which uses protein databases to compare to experimental masses obtained. The mass spectrometry data are plotted in Fig. 2e, denoting the peptide fragments specific to sFRP4.
Formation of secondary structures of any recombinant protein is crucial for retention of its functionality. After the rigorous process of purification with detergents and subsequent dialysis, involving denaturation, renaturation, and refolding steps, it was essential to know the integrity of secondary structure. Hence, purified and dialyzed GST-sFRP4 was subjected to circular dichroism spectral analysis, which generated a spectrum of scanning wavelength versus millidegree. Figure 2f depicts the graph of scanning wavelength versus mean residue ellipticity (mdeg cm2 dmol−1), which was obtained by the following formula:
here, θ is ellipticity in millidegree, C is concentration of protein, n gives number of residues, and l gives the path length [43].
The percentages of α-helices and β-sheets, as determined by applying Yang’s algorithm, were found to be 20 and 11.9 %, respectively.
Western blotting with anti-GST antibody confirmed the presence of GST-tagged sFRP4 at the legitimate position corresponding to 64 kDa of GST-sFRP4. As a control experiment, GST protein purified from pGEX-4T2 bearing E. coli BL21 cells, was also blotted (supporting information Fig S1).
Prediction of 3D structure of GST-sFRP4 and its docking with Wnt7a
Docking studies were carried out for GST-sFRP4 with one of the Wnt ligands known to bind to sFRP4 [30]. The in silico study was performed as there was no report available on the functionality of the GST-tagged sFRP4 bound with Wnt ligands. First, the 3D structures of CRD (binding domain) of sFRP4 (henceforth referred to as CRD-sFRP4, supporting information Fig S2A), sFRP4 (Fig S2B), GST-sFRP4 (Fig S2C), and Wnt7a (Fig S2D) were predicted due to lack of the reports on crystal structures of sFRP4 and Wnts. It should be mentioned here that we have previously reported the predicted structure of Wnt7a [3]. The only available crystal structure of sFRP superfamily is that of CRD of sFRP3 (PDB ID: 1IJXA) [25]. Therefore, CRD of sFRP3 was automatically detected by Phyre2 server as a template for predicting the structure of CRD of sFRP4. 3D structure of Wnt7a was generated with I-Tasser prediction server. Validation of the predicted structures was done by aligning CRD-sFRP4 with the CRD of sFRP3 in PyMol, which generated a root mean square deviation (RMSD) value of 0.326 (supporting information Fig S2E), indicating nearly similar structural folds. The same comparison was done between CRD-sFRP4 and GST-sFRP4 structures, which generated an RMSD value of 0.584 (supporting information Fig S2F), respectively. Both cases demonstrated that binding domain remained intact in the predicted structures of sFRP4 and GST-sFRP4, as shown by negligible values of RMSD. Moreover, the lower RMSD between CRD-sFRP4 and GST-sFRP4 suggested that tagging of GST to sFRP4 slightly changed the protein conformation in favor of binding. This may be explained on the basis of previous studies, where tagging of GST to recombinant proteins helps to stabilize them [44]. After confirming the validity of the binding domain, which is primarily responsible for binding of Wnt ligands, the structures were docked with Wnt. Each of the CRD-sFRP4, sFRP4, and GST-sFRP4 was separately docked with Wnt7a using ClusPro (Fig. 3a–c, respectively). Analysis of interactions between the docked structures of lowest binding energies was done using PDBsum Generate (supporting information Fig S3A, S3B, and S3C).

Docked structures of Wnt7a with a CRD-sFRP4, b sFRP4, c GST-sFRP4. Docking was done using ClusPro protein–protein docking server. MTT assay depicting dose-dependent anti-proliferative effect of GST-sFRP4 d on HeLa cells and e A549 cells
Anti-proliferative effect of GST-sFRP4
The Wnt signal transduction is known to be responsible for aberrant proliferation in cancer cells, where inhibitory proteins like sFRP4 are hypermethylated. Although there are a few reports demonstrating that transfection of sFRP4 antagonizes the Wnt pathway, to the best of our knowledge there has been no documentation of the therapeutic effect of bacterially expressed recombinant sFRP4. In our study, MTT assays revealed that exogenous addition of GST-sFRP4 to HeLa and A549 cancer cells significantly inhibited cell proliferation in a dose-dependent fashion (Fig. 3d, e, respectively). HeLa cells when treated with up to 32 nM of protein for 48 h resulted in reduction of viable cells to below 60 %. In case of A549 cells, a similar effect was seen at a maximum concentration of 20 nM of recombinant protein. Postdialysis buffer controls were kept for all MTT assays, which confirmed the complete removal of contaminating agents from the purified protein fractions, such as L-reduced glutathione and detergents (data not shown).
Targeting the classical Wnt/β-catenin signaling with GST-sFRP4
The complex nature of signaling pathways and their intersection with a plethora of other pathways often makes it extremely difficult to decipher their functions. Whether the family of sFRPs always targets the canonical Wnt pathway is a debatable issue, since there have been several reports stating the contrary. Nearly all of the sFRPs, including sFRP4, have been shown to behave in a conflicting manner [17, 18, 45, 46]. In order to understand the underlying mechanism of cell growth inhibition observed in the cell viability assays and to shed some light on the implicated pathway, we probed the expression of certain critical members of the canonical Wnt pathway. One such central molecule is the β-catenin, which has a stable cytosolic expression during Wnt signal transduction and promotes the upregulation of downstream pro-proliferative genes, such as cyclin D1, survivin, and c-myc. However, when the pathway is shut down due to the presence of some inhibitory molecule, β-catenin is phosphorylated and thus marked for degradation, thereby blocking the transcription of the downstream genes. Therefore, we checked the expression levels of total β-catenin as well as its phosphorylated form in treated and untreated HeLa and A549 cells. After quantifying total protein content of each sample by Lowry assay, 60 µg of protein was loaded in each well. In both cell types, expression of β-catenin was greatly reduced in comparison with control experiments. Specifically in case of HeLa, treated cells possessed negligible amounts of β-catenin protein. When probed with antibody specific for the Ser33/Ser37 phosphorylated form of β-catenin, expression was substantially increased in treated cells, with respect to the untreated control cells. Ratio of phosphorylated to total β-catenin increased greatly in sFRP4-treated HeLa cells in comparison with control cells (Fig. 4a). Furthermore, expressions of cyclin D1, survivin, and c-myc genes were analyzed by real-time PCR. In HeLa cells, expression of all of the above three genes was reduced approximately twofold after treatment with GST-sFRP4, with respect to untreated cells (Fig. 4b). This is in accordance with the above Western blot analysis data, which demonstrated that β-catenin was degraded on treatment with GST-sFRP4 and was thus unable to transcribe the downstream genes. It can be inferred from these expression analyses that treatment with GST-sFRP4 resulted in antagonizing of the Wnt, thereby blocking downstream signaling leading to phosphorylation and subsequent ubiquitination of β-catenin. In A549 cells (Fig. 4c, d), a similar trend was observed. Ratio of phosphorylated to total β-catenin increased significantly in sFRP4-treated A549 cells in comparison with the untreated cells. Expression of survivin gene was found to decrease slightly (approximately 1.3-fold) and expression of c-myc was reduced nearly twofold on treatment with recombinant protein. However, treatment failed to induce changes in the levels of cyclin D1 gene. All values were normalized with respect to expression of β-actin gene. These data provided evidence of β-catenin-dependent regulation of the canonical Wnt pathway upon GST-sFRP4 treatment.

Expression analysis of key players of the classical Wnt pathway. a Western blotting showed that ratio of phosphorylated to total β-catenin increased significantly in sFRP4-treated HeLa cells with respect to control. b Real-time PCR analysis showed downstream genes cyclin D1, c-myc, and survivin genes were repressed in treated cells. c Western blotting of control and treated A549 cells revealed that ratio of phospho-β-catenin to total β-catenin increased in treated cells. d Real-time PCR analysis of A549 cells showed downregulation of c-myc and survivin genes with respect to untreated cells, while cyclin D1 remained nearly unaltered. Each bar represents the fold change in expression of a particular gene (cyclin D1, c-myc, or survivin) in sFRP4 treated cells with respect to untreated cells with a set value of 1
Efficacy of co-therapy
In our earlier experiment to assess cell viability of sFRP4-treated cells, we found that increasing the concentration or time of treatment did not lead to any further enhancement of anti-proliferative effect. This encouraged us to test the potential of the recombinant sFRP4 to chemosensitize cancer cells toward conventional anticancer drugs, in order to augment the efficacy of recombinant sFRP4. Hence, we designed a module of co-therapy of sFRP4 with conventional anti-tumor drugs, namely cisplatin and doxorubicin. MTT assays showed a significant increase in enhancement of efficacy in case of combination therapy as compared to either the protein or the drug alone. This chemosensitizing effect of recombinant protein would help to greatly minimize the adverse side effects of the drugs, when implemented clinically. In HeLa cells, 3 nM of protein was sufficient to reduce the percentage of viable cells from 90 % in case of only 4 μM cisplatin to 40 % in case of combination therapy (Fig. 5a). A similar scenario was observed for A549 cells, where 20–30 % reduction of viable cells was observed in combination with protein (Fig. 5b). In the other instance of doxorubicin (Fig. 5c, d for HeLa and A549, respectively), cell viability was reduced by about 20 % when sFRP4 was added to the treatment module, even for very low concentration of doxorubicin (0.1–0.6 μM). These results led us to understand that this mode of co-therapy would be a possible choice for futuristic cancer therapy with minimum side effects.

Cell viability assay after 48 h of combination therapy, a HeLa cells treated with increasing concentration of cisplatin and 20 nM of GST-sFRP4, b A549 cells treated with increasing concentration of cisplatin and 12 nM of GST-sFRP4, c HeLa cells treated with increasing concentration of doxorubicin and 3 nM of GST-sFRP4, d A549 cells treated with increasing concentration of doxorubicin and 8 nM of GST-sFRP4
Determination of cell cycle arrest by flow cytometry
To substantiate the above findings, the mode of anti-proliferative effect was investigated by flow cytometric analysis and dual staining methods. FACS-based analysis was performed to ascertain the effect of recombinant sFRP4, alone and in combination with chemotherapeutic drugs on the cell cycle using PI. A low concentration of sFRP4 (12 nM) was found to be sufficient to induce arrest in G2/M phase for both cell lines (15–34 % in HeLa and 10.19–21 % in A549) in 48 h. However, in case of A549 cells, there was a significant increase in cells undergoing S phase arrest (5 % in control to 16 % in treated). A low range of protein concentration was selected to demonstrate the augmented effect of co-therapy. In combination of cisplatin with GST-sFRP4, both cells showed an increase in percentage of cells in G2/M phase by approximately 12 %, in comparison with cisplatin alone. For HeLa, 50 % of cells exhibited G2/M arrest on this mode of co-therapy, whereas the percentage was 67 % for A549. In case of co-therapy with doxorubicin, differential behavior was observed for the two cell types. While HeLa cells exhibited S phase arrest, A549 cells showed G2/M blocking of cell cycle. The cell cycle analysis is depicted in Fig. 6a, b, for HeLa and A549 cells, respectively. Graphical representation of the same is given in the supporting information (Fig S4).


Flow cytometry-based cell cycle analysis of untreated and treated a HeLa cells, b A549 cells
Induction of apoptosis in a co-therapy module
Onset of apoptosis was determined by exploiting its underlying mechanism by means of Annexin V-FITC/PI dual staining using flow cytometry. Labeled Annexin V is an indicator of translocation of phosphatidylserine from inner to outer leaflet of cell membrane, a characteristic event of apoptosis. It labels both early and late apoptotic cells till the membrane is completely disintegrated. On the other hand, PI is a membrane impermeable dye, which can intercalate with nucleic acids only when membrane is damaged. Hence, this system was employed to quantitatively ascertain the percentages of viable (Annexin V−, PI−), early apoptotic (Annexin V+, PI−), late apoptotic (Annexin V+, PI+), very late apoptotic/necrotic (Annexin V−, PI+) cells by flow cytometric analysis. Percentage of live cells was reduced from 91.4 to 73 % for HeLa cells and 92.5–82.8 % for A549 cells after 48 h of treatment with a very low concentration (12 nM) of GST-sFRP4 (supporting information Fig S5 and S6 for HeLa and A549 cells, respectively); early apoptotic cells were found to increase by 16 % for HeLa cells, whereas for A549 there was a marginal increase in each quadrant depicting apoptotic cells. Co-therapy resulted in an increase of late apoptotic cell population. Graphical representation of the same is given in the supporting information (Fig S7). Apoptosis was also detected by bivariate analysis of dual stained (AO/EB) cancer cells, details of which are provided in the supporting information (Fig S8A and S8B). Hence, it was established that recombinant sFRP4 as well as combination therapy with cisplatin/doxorubicin induced an apoptotic mode of cell death. This was in accordance with previous reports delineating the mode of action of sFRP4 in keratinocytes [14] and breast cancer cells [47].
Quantitative expression profiling of Wnt downstream genes by real-time PCR
To address the mechanism underlying the successful application of co-therapy, we checked the quantitative expressions of pro-proliferative genes cyclin D1, c-myc, and survivin by quantitative real-time PCR analysis. While all three genes are known to be regulated by the Wnt/β-catenin pathway, their behavior after treatment with combination therapy is unknown. During our investigation, we observed that in 24 h, cyclin D1 was transcriptionally downregulated by 30 and 10 % on treatment with cisplatin alone, in HeLa and A549 cells, respectively (Fig. 7a, b, respectively). However, in both cell types, there was a significant (~50 %) difference in expression of cyclin D1 between treatment with cisplatin alone, and its combination with GST-sFRP4, indicating a further inhibition of cell proliferation. From this, we concluded that the recombinant sFRP4 inhibited the Wnt pathway even in the presence of potent anticancer drug cisplatin. Also, expression of cyclin D1 was substantially decreased in case of co-therapy, as opposed to either mode of treatment alone. In case of therapy with doxorubicin alone, HeLa cells (Fig. 7a) exhibited reduced levels of cyclin D1 (by 50 %). Although there was a further decrease in expression level on co-therapy with protein, it was marginal, possibly because even in low concentrations, doxorubicin itself is potent enough to substantially inhibit cell proliferation. However for A549 cells, there was a distinct reduction (by nearly 40 %) in case of co-therapy, as compared to doxorubicin alone (Fig. 7b). In addition to cyclin D1, the expression of c-myc gene was also probed (Fig. 7c, d for HeLa and A549, respectively). Treatment with cisplatin alone resulted in nearly 20 % decrease in the level of c-myc in both cells, compared to untreated cells. In HeLa, this expression was further reduced by 30 % on co-therapy of cisplatin with GST-sFRP4; whereas in case of A549 cells, co-therapy brought about a further reduction by 60 % compared to treatment with only cisplatin. Doxorubicin treatment yielded a similar effect on c-myc as seen for cyclin D1. Treatment with drug alone lowered gene expression to 60 and 20 %, with respect to control, for HeLa and A549 cells, respectively. In both cases, there was a further reduction of approximately 20 % for co-therapy of doxorubicin with recombinant sFRP4. Lastly, the expression of survivin gene was determined (Fig. 7e, f for HeLa and A549, respectively). Both the chemotherapeutic drugs had an immense effect in decreasing the levels of survivin. Therefore, the effect of co-therapy on survivin expression was observed to be marginal in both cell lines. These quantitative data revealed that while the recombinant sFRP4 as well as conventional anticancer agents individually led to reduced proliferation of cells, their synergistic effect proved to be significantly more fruitful.

Real-time PCR expression analysis of untreated and treated cells, a cyclin D1 in HeLa, b cyclin D1 in A549, c c-myc in HeLa, d c-myc in A549, e survivin in HeLa, f survivin in A549
Discussion
In this investigation, we cloned the sFRP4 from a hitherto unknown source—ACHN renal carcinoma cell line. ACHN cells possess a low level of expression of sFRP4, which compelled us to perform re-PCR in order to acquire sufficient gene product for cloning. Rigorous solubilization and purification process yielded recombinant sFRP4 ready for application. In a previous report, we had demonstrated the therapeutic application of bacterially expressed GST-tagged sFRP1 [3, 48]. Bolstered by those results, we designed this study with a similar, yet more mechanistic approach.
Before proceeding to experiment with the GST-tagged sFRP4, we screened a few cell lines of different origins, to ensure that Wnt ligands were expressed. The gene profile obtained in our results was found to be in agreement with previous reports documenting the expression of Wnt ligands in cancer. In addition, docking studies revealed that tagging with GST seemed to improve its stability of binding to Wnt ligand. Although there is no report stating that GST-tagged protein displays improved binding properties with target ligands, GST is known to enhance stability of the tagged protein [49], which in turn could be responsible for its improved binding capacity.
Equipped with the evidence gathered from these data, a holistic approach was assumed in establishing the functionality of recombinant sFRP4 in blocking the Wnt signaling. We showed that the decrease in viability exhibited by HeLa and A549 cells was due to inhibition of the Wnt pathway by recombinant sFRP4. Although MALDI and circular dichroism results demonstrated the authenticity and intact secondary structure formation of the bacterially expressed GST-sFRP4, blocking of the Wnt cascade proved beyond doubt that the protein retained its functionality even after extensive extraction and purification steps. We speculate that the use of Triton X-100 to capture sarkosyl in its micelle during solubilization of protein as well as subsequent stepwise dialysis helped in the refolding of the recombinant sFRP4. Although it is plausible that expressing of the recombinant sFRP4 using yeast may show better anti-proliferative effect due to protein folding analogous to that in mammalian cells, but functional expression in bacterial host in the present study is an easy approach for generation of recombinant sFRP4.
While there are three reported pathways of Wnt activation, the canonical β-catenin-dependent signaling has often been implicated in cancer formation and progression. However, there have been contradictory reports stating otherwise [17, 18]. Therefore, we validated the involvement of the Wnt/β-catenin pathway by analyzing key players of the downstream pathway by Western blotting or quantitative real-time PCR analysis. Activation of the signaling in most forms of cancer leads to stabilization of free pools of β-catenin in the cytoplasm. A similar observation was made by Western blotting of total protein from untreated HeLa and A549 cells. Translocation of the stabilized β-catenin into the nucleus activated transcription of pro-proliferative target genes, such as cyclin D1, c-myc, and survivin, as shown by quantitative real-time PCR. Binding of recombinant sFRP4 with corresponding Wnt ligands prevented the signal transduction, which led to phosphorylation and subsequent ubiquitination of β-catenin, as documented by Western blotting. Correspondingly, downregulation of downstream genes, namely cyclin D1, c-myc, and survivin, was observed by real-time PCR. These experiments proved that recombinant sFRP4 executed its anti-proliferative activity by binding to Wnts and blocking the downstream signal cascade. However, treatment with traditional anti-tumor drugs, cisplatin and doxorubicin also suppressed expression of these genes. As there is no known link directly connecting Wnt cascade and these drugs, we hypothesize that the drugs could be inhibiting these oncogenes via other signaling pathways. For instance, cisplatin has been reported to induce apoptosis mediated by p53 [50, 51], which in turn is known to suppress cyclin D1 [52] as well as c-myc [53]. Cisplatin is also known to be implicated in Ras/Akt/ERK pathways, which have cyclin D1 and c-myc as their downstream targets [54]. Survivin belongs to the inhibitor of apoptosis (IAP) protein family and has been reported to be suppressed by p53 [55]. Doxorubicin, an inhibitor of topoisomerase II, has been known to downregulate cyclin D1 [56], c-myc [57–59], and survivin [60, 61]. However, it should be noted here that signaling pathways are complicated, interlinked, obscure, and often behave in conflicting manner. Hence, detailed mechanism of action by cisplatin or doxorubicin was beyond the scope of this study. Herein, we established that recombinant sFRP4 repressed the expression of key members of the Wnt pathway involved in aberrant proliferation of cancer cells. It should also be mentioned here that significant enhancement of inhibition of these oncogenes was observed after combination therapy, as compared to individual treatment modality. Notably, effect of sFRP4 in blocking of the Wnt signaling was not violated even in course of combination module.
The mode of cell inhibition was determined by flow cytometric and microscopic analyses, which demonstrated cell cycle arrest followed by apoptotic cell death. Programmed cell death is called apoptosis, which is a more desirable form of cell elimination than necrosis. It is conceivable that treatment of HeLa and A549 cells with GST-sFRP4 as well as combination therapy with drugs induced an appreciable arrest in the G2/M phase of cell cycle. This would also be a logical explanation for inhibition of cell proliferation and for the subsequent apoptosis, which was shown to be mediated by the regulation of cell cycle. This was consistent with previous reports defining the mode of cell death by sFRP4 [14, 47]. Use of low concentration of protein for FACS-based analyses may be correlated with the percentage of apoptotic population (20–27 %) in case of treatment with protein alone. Also, the fact that sFRP4 affected the cells by holding them in a steady-state population possibly brought about chemosensitization to cisplatin and doxorubicin. Individually, each of the chemotherapeutic drugs induced an arrest in the G2/M phase of the cycle, consistent with previous reports [56]. In cancer, many cell cycle checkpoints, including G2/M checkpoints, are defective, resulting in continuous proliferation and a propensity to acquire chromosomal aberrations causing drug resistance [62]. By causing G2/M arrest, recombinant sFRP4 created an externally imposed cell cycle checkpoint. This not only caused apoptotic cell death by itself, but also made the cells susceptible to chemotherapy, thus ensuring that a very minimal concentration of drug was sufficient to produce massive apoptosis. When applied clinically, this would be advantageous in curtailing the undesirable side effects of these drugs. Further detailed studying of the molecular mechanisms involved in the dynamics of this pathway will pave the way for its clinical usage. Recently, Wnt pathway inhibitors have been studied successfully to block Wnt signaling in mouse models [63, 64]. From this, it may be inferred that the bacterially expressed recombinant sFRP4 holds immense potential for in vivo cancer therapy.
Conclusions
Attempting to implement the existing literature on Wnt to concoct a novel approach of employing bacterially expressed recombinant human protein to inhibit this signaling pathway, we cloned human sFRP4 and purified the GST-tagged protein from bacterial host. The proficiency displayed by recombinant sFRP4 in impeding the Wnt signaling may open up new possibilities for recombinant protein therapeutics. The functionality demonstrated by the bacterially expressed human sFRP4 could also be exploited for studying its interactions with various Wnt isoforms. It could also be used to resolve the discrepancies regarding blocking of canonical or non-canonical or any other intersecting signaling due to binding of sFRP4 to different Wnt ligands. Further, efficacy of the treatment module was augmented significantly by co-therapy with traditional chemotherapeutic drugs. The approach of compiling the benefits of recombinant protein therapy to lower the dosage of drugs has the potential to minimize side effects and circumvent the development of resistance, making it a tremendously fascinating solution for tackling a complex disease like cancer. The highlights of this investigation encompass the identification and possible application of a novel mode of recombinant protein therapeutics.
Abbreviations
- sFRP:
-
Secreted frizzled-related protein
- GST:
-
Glutathione S-transferase
- CRD:
-
Cysteine-rich domain
- NLD:
-
Netrin-like domain
- MALDI TOF:
-
Matrix-assisted laser desorption/ionization time of flight
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- cDNA:
-
Complementary deoxyribonucleic acid
- RNA:
-
Ribonucleic acid
- IPTG:
-
Isopropyl β-d-1-thiogalactopyranoside
- PBS:
-
Phosphate-buffered saline
- PMSF:
-
Phenylmethylsulfonyl fluoride
- EDTA:
-
Ethylenediaminetetraacetic acid
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- HRP:
-
Horseradish peroxidase
- HCl:
-
Hydrochloric acid
- PI:
-
Propidium iodide
- FACS:
-
Fluorescence-activated cell sorting
- IAP:
-
Inhibitor of apoptosis
- FITC:
-
Fluorescein isothiocyanate
- AO:
-
Acridine orange
- EB:
-
Ethidium bromide
References
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13:11–26
Huang S, Zhong X, Gao J, Song R, Wu H, Zi S, Yang S, Du P, Cui L, Yang C, Li Z (2014) Coexpression of SFRP1 and WIF1 as a prognostic predictor of favorable outcomes in patients with colorectal carcinoma. Biomed Res Int 2014:256723
Ghoshal A, Ghosh SS (2015) Expression, purification, and therapeutic implications of recombinant sFRP1. Appl Biochem Biotechnol 175:2087–2103
Dow Lukas E, O’Rourke Kevin P, Simon J, Tschaharganeh Darjus F, van Es Johan H, Clevers H, Lowe Scott W (2015) Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 161:1539–1552
Pohl S, Scott R, Arfuso F, Perumal V, Dharmarajan A (2015) Secreted frizzled-related protein 4 and its implications in cancer and apoptosis. Tumour Biol 36:143–152
Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB (2002) A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31:141–149
Carmen J, Marsit MRK, Andrew Angeline et al (2005) Epigenetic inactivation of SFRP genes and TP53 alteration act jointly as markers of invasive bladder cancer. Cancer Res 65:7081–7085
Lee AY, He B, You L, Dadfarmay S, Xu Z, Mazieres J, Mikami I, McCormick F, Jablons DM (2004) Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene 23:6672–6676
Brebi P, Hoffstetter R, Andana A, Ili CG, Saavedra K, Viscarra T, Retamal J, Sanchez R, Roa JC (2014) Evaluation of ZAR1 and SFRP4 methylation status as potentials biomarkers for diagnosis in cervical cancer: exploratory study phase I. Biomarkers 19:181–188
Jacob F, Ukegjini K, Nixdorf S, Ford CE, Olivier J, Caduff R, Scurry JP, Guertler R, Hornung D, Mueller R, Fink DA, Hacker NF, Heinzelmann-Schwarz VA (2012) Loss of secreted frizzled-related protein 4 correlates with an aggressive phenotype and predicts poor outcome in ovarian cancer patients. PLoS ONE 7:e31885
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398:422–426
Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, Boman BM (2001) Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 61:8664–8667
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281:1509–1512
Maganga R, Giles N, Adcroft K, Unni A, Keeney D, Wood F, Fear M, Dharmarajan A (2008) Secreted frizzled related protein-4 (sFRP4) promotes epidermal differentiation and apoptosis. Biochem Biophys Res Commun 377:606–611
Dharmarajan A, McLaren S, White E, Zeps N (2006) Expression of secreted frizzled related protein-4 (sFRP-4) and associated Wnt signalling in breast cancer. Cancer Res 66:1238
Lisa G, Horvath SMH, Kench James G et al (2004) Membranous expression of secreted frizzled-related protein 4 predicts for good prognosis in localized prostate cancer and inhibits PC3 cellular proliferation in vitro. Clin Cancer Res 10:615–625
Surana R, Sikka S, Cai W, Shin EM, Warrier SR, Tan HJ, Arfuso F, Fox SA, Dharmarajan AM, Kumar AP (2014) Secreted frizzled related proteins: implications in cancers. Biochim Biophys Acta 1845:53–65
Huang D, Yu B, Deng Y, Sheng W, Peng Z, Qin W, Du X (2010) SFRP4 was overexpressed in colorectal carcinoma. J Cancer Res Clin Oncol 136:395–401
Saran U, Arfuso F, Zeps N, Dharmarajan A (2012) Secreted frizzled-related protein 4 expression is positively associated with responsiveness to cisplatin of ovarian cancer cell lines in vitro and with lower tumour grade in mucinous ovarian cancers. BMC Cell Biol 13:25
Warrier S, Balu SK, Kumar AP, Millward M, Dharmarajan A (2013) Wnt antagonist, secreted frizzled-related protein 4 (sFRP4), increases chemotherapeutic response of glioma stem-like cells. Oncol Res 21:93–102
Longman D, Arfuso F, Viola HM, Hool LC, Dharmarajan AM (2012) The role of the cysteine-rich domain and netrin-like domain of secreted frizzled-related protein 4 in angiogenesis inhibition in vitro. Oncol Res 20:1–6
Lopez-Rios J, Esteve P, Ruiz JM, Bovolenta P (2008) The netrin-related domain of Sfrp1 interacts with Wnt ligands and antagonizes their activity in the anterior neural plate. Neural Dev 3:19
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG, Baylin SB (2004) Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 36:417–422
Jones SE, Jomary C (2002) Secreted frizzled-related proteins: searching for relationships and patterns. BioEssays 24:811–820
Dann CE, Hsieh JC, Rattner A, Sharma D, Nathans J, Leahy DJ (2001) Insights into Wnt binding and signalling from the structures of two frizzled cysteine-rich domains. Nature 412:86–90
Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738
Roy A, Yang J, Zhang Y (2012) COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40:W471–W477
Carmon KS, Loose DS (2008) Secreted frizzled-related protein 4 regulates two Wnt7a signaling pathways and inhibits proliferation in endometrial cancer cells. Mol Cancer Res 6:1017–1028
Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, Vajda S (2013) How good is automated protein docking? Proteins 81:2159–2166
Kozakov D, Brenke R, Comeau SR, Vajda S (2006) PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65:392–406
Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50
Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res 32:W96–W99
Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM (1997) PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci 22:488–490
Laskowski RA (2001) PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res 29:221–222
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461:614–620
Schwanke RC, Renard G, Chies JM, Campos MM, Batista EL Jr, Santos DS, Basso LA (2009) Molecular cloning, expression in Escherichia coli and production of bioactive homogeneous recombinant human granulocyte and macrophage colony stimulating factor. Int J Biol Macromol 45:97–102
Singh A, Upadhyay V, Panda A (2015) Solubilization and refolding of inclusion body proteins. In: García-Fruitós E (ed) Insoluble proteins. Springer, New York, pp 283–291
Mercado-Pimentel ME, Jordan NC, Aisemberg GO (2002) Affinity purification of GST fusion proteins for immunohistochemical studies of gene expression. Protein Expr Purif 26:260–265
Yip CK, Kimbrough TG, Felise HB, Vuckovic M, Thomas NA, Pfuetzner RA, Frey EA, Brett Finlay B, Miller SI, Strynadka NCJ (2005) Structural characterization of the molecular platform for type III secretion system assembly. Nature 435:702–707
Gattiker A, Bienvenut WV, Bairoch A, Gasteiger E (2002) FindPept, a tool to identify unmatched masses in peptide mass fingerprinting protein identification. Proteomics 2:1435–1444
Achilonu I, Siganunu TP, Dirr HW (2014) Purification and characterisation of recombinant human eukaryotic elongation factor 1 gamma. Protein Expr Purif 99C:70–77
Terpe K (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 60:523–533
Joesting MS, Perrin S, Elenbaas B, Fawell SE, Rubin JS, Franco OE, Hayward SW, Cunha GR, Marker PC (2005) Identification of SFRP1 as a candidate mediator of stromal-to-epithelial signaling in prostate cancer. Cancer Res 65:10423–10430
Qu Y, Ray PS, Li J, Cai Q, Bagaria SP, Moran C, Sim MS, Zhang J, Turner RR, Zhu Z, Cui X, Liu B (2013) High levels of secreted frizzled-related protein 1 correlate with poor prognosis and promote tumourigenesis in gastric cancer. Eur J Cancer 49:3718–3728
Dharmarajan A, Zeps N, McLaren S (2005) Expression of secreted frizzled related protein-4 (sFRP-4) and associated Wnt signalling in cancer and apoptosis. Reprod Fertil Dev 17:63
Ghoshal A, Goswami U, Sahoo AK, Chattopadhyay A and Ghosh SS (2015) Targeting Wnt canonical signaling by recombinant sFRP1 bound luminescent Au-nanocluster embedded nanoparticles in cancer theranostics. ACS Biomater Sci Eng 1:1256–1266
Park DW, Kim SS, Nam MK, Kim GY, Kim J, Rhim H (2011) Improved recovery of active GST-fusion proteins from insoluble aggregates: solubilization and purification conditions using PKM2 and HtrA2 as model proteins. BMB Rep 44:279–284
Bragado P, Armesilla A, Silva A, Porras A (2007) Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 12:1733–1742
Ikeguchi M, Tatebe S, Kaibara N, Ito H (1997) Changes in levels of expression of p53 and the product of the bcl-2 in lines of gastric cancer cells during cisplatin-induced apoptosis. Eur Surg Res 29:396–402
Rocha S, Martin AM, Meek DW, Perkins ND (2003) p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol Cell Biol 23:4713–4727
Ho JS, Ma W, Mao DY, Benchimol S (2005) p53-dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol 25:7423–7431
Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4:257–262
Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, Wen SF, Wang L, Kirschmeier P, Bishop WR, Nielsen LL, Pickett CB, Liu S (2002) Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21:2613–2622
Rezaei PF, Fouladdel S, Ghaffari SM, Amin G, Azizi E (2012) Induction of G1 cell cycle arrest and cyclin D1 down-regulation in response to pericarp extract of Baneh in human breast cancer T47D cells. DARU J Pharm Sci 20:1–5
Pourquier P, Montaudon D, Huet S, Larrue A, Clary A, Robert J (1998) Doxorubicin-induced alterations of c-myc and c-jun gene expression in rat glioblastoma cells: role of c-jun in drug resistance and cell death. Biochem Pharmacol 55:1963–1971
Clary A, Larrue A, Pourquier P, Robert J (1998) Transcriptional down-regulation of c-myc expression in an erythroleukemic cell line, K562, and its doxorubicin-resistant variant by two topoisomerase II inhibitors, doxorubicin and amsacrine. Anticancer Drugs 9:245–254
Fornari FA Jr, Jarvis WD, Grant S, Orr MS, Randolph JK, White FK, Mumaw VR, Lovings ET, Freeman RH, Gewirtz DA (1994) Induction of differentiation and growth arrest associated with nascent (nonoligosomal) DNA fragmentation and reduced c-myc expression in MCF-7 human breast tumor cells after continuous exposure to a sublethal concentration of doxorubicin. Cell Growth Differ 5:723–733
Lee BS, Kim SH, Jin T, Choi EY, Oh J, Park S, Lee SH, Chung JH, Kang SM (2013) Protective effect of survivin in doxorubicin-induced cell death in h9c2 cardiac myocytes. Korean Circ J 43:400–407
Estève P-O, Chin HG, Pradhan S (2007) Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J Biol Chem 282:2615–2625
Ahmad N, Feyes DK, Agarwal R, Mukhtar H, Nieminen A-L (1997) Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst 89:1881–1886
Wickstrom M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjornsson B, Sanden E, Darabi A, Siesjo P, Kool M, Kogner P, Baryawno N and Johnsen JI (2015) Wnt/[beta]-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat Commun 6:8904
Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, Fischer M, Chaudhari A, Ji M, Kapoun AM, Lam A, Lazetic S, Ma S, Mitra S, Park I-K, Pickell K, Sato A, Satyal S, Stroud M, Tran H, Yen W-C, Lewicki J, Hoey T (2012) Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci 109:11717–11722
Acknowledgments
Authors acknowledge financial supports from the Department of Biotechnology Programme Support (BT/PR13560/COE/34/44/2015) and the Department of Electronics and Information Technology [No. 5(9)/2012-NANO (Vol. II)]. Authors also acknowledge the support of the Centre for Nanotechnology and the Central Instruments Facility, IIT Guwahati.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghoshal, A., Ghosh, S.S. Antagonizing canonical Wnt signaling pathway by recombinant human sFRP4 purified from E. coli and its implications in cancer therapy. Mol Cell Biochem 418, 119–135 (2016). https://doi.org/10.1007/s11010-016-2738-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2738-6