
Abstract
Post-traumatic hypertrophic scar (HS) is a fibrotic disease with excessive extracellular matrix (ECM) production, which is a response to tissue injury by fibroblasts. Although emerging evidence has indicated that miRNA contributes to hypertrophic scarring, the role of miRNA in HS formation remains unclear. In this study, we found that miR-143-3p was markedly downregulated in HS tissues and fibroblasts (HSFs) using qRT-PCR. The expression of connective tissue growth factor (CTGF/CCN2) was upregulated both in HS tissues and HSFs, which is proposed to play a key role in ECM deposition in HS. The protein expression of collagen I (Col I), collagen III (Col III), and α-smooth muscle actin (α-SMA) was obviously inhibited after treatment with miR-143-3p in HSFs. The CCK-8 assay showed that miR-143-3p transfection reduced the proliferation ability of HSFs, and flow cytometry showed that either early or late apoptosis of HSFs was upregulated by miR-143-3p. In addition, the activity of caspase 3 and caspase 9 was increased after miR-143-3p transfection. On the contrary, the miR-143-3p inhibitor was demonstrated to increase cell proliferation and inhibit apoptosis of HSFs. Moreover, miR-143-3p targeted the 3′-UTR of CTGF and caused a significant decrease of CTGF. Western blot demonstrated that Akt/mTOR phosphorylation and the expression of CTGF, Col I, Col III, and α-SMA were inhibited by miR-143-3p, but increased by CTGF overexpression. In conclusion, we found that miR-143-3p inhibits hypertrophic scarring by regulating the proliferation and apoptosis of human HSFs, inhibiting ECM production-associated protein expression by targeting CTGF, and restraining the Akt/mTOR pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertrophic scar (HS) is a dermal fibroproliferative disorder which usually develops during the healing process after skin trauma or severe burn injury [1]. It is characterized by excessive extracellular matrix (ECM) deposition, including collagen, elastin, α-smooth muscle actin (α-SMA), fibronectin, and matrix-directed proteases and protease inhibitors [2–4]. This causes a deformed appearance and contracted neogenic tissue, leading to a serious danger to the physical and psychological health of patients. Presently, several methods have been used to prevent and cure hypertrophic scars, such as surgical excision, radiation therapy, and steroid injections, but none have proven to be optimal, and the clinical behavior of hypertrophic scarring remains unclear. Some research has shown that many different non-coding RNA and growth factors are involved in the formation of hypertrophic scars [5, 6].
MicroRNAs (miRNAs) are short (19–24 nt) non-coding RNAs that play critical roles in many important biological processes, such as cancerogenesis [7] and fibroblasts activation [8]. They control target mRNA translation and stability by binding to regulatory sites that are mostly located in the 3′-untranslated region (UTR) of transcripts [9]. Furthermore, aberrant expression of miRNAs has been associated with several pathological processes, including fibrosis and ECM metabolism in organs including the heart, kidneys, liver, and lungs [10]. Recently, some miRNAs have been reported to contribute to hypertrophic scarring or keloid formation, and differentially expressed miRNAs have been identified through genomic profiling between denatured dermis and normal skin (NS), implicating multiple signaling pathways in wound healing [11]. In particular, the miR-29 family members (miR-29a, miR-29b, and miR-29c) have been reported to directly regulate the translation of various ECM mRNAs, such as the collagen superfamily [12]. Thus, more studies are needed to further validate the functional roles of miRNAs and relevant signaling pathway in the pathogenesis of hypertrophic scarring.
Connective tissue growth factor (CTGF/CCN2) is a matricellular protein which plays an important role in promoting fibrosis and scarring in numerous tissues [13, 14]. Overexpression of CTGF has been shown to promote fibrosis and scar formation in the skin, kidney, liver, brain, and lung [13]. Baseline CTGF expression is increased in unstimulated HS fibroblasts, as compared to that in normal fibroblasts. Furthermore, CTGF has been reported to mediate the ability of TGF-β1 to stimulate ECM synthesis, indicating that CTGF plays a critical role in the regulation of cartilage development and ECM production [15]. On the other side, it has been reported that both miR-133 and miR-30 directly downregulate CTGF in models of heart failure [16], thereby establishing an important role for miRNAs in the control of structural changes in the ECM. To our knowledge, the regulation of CTGF by microRNAs (miRNAs) has not yet been described in hypertrophic scarring. CTGF is capable of activating the ERK, JNK, and p38 pathways [17]. In lung fibrosis, CTGF activates the promoter of collagen type I α2 by a mechanism that is dependent on the ERK-1/2 and JNK MAPK pathways [18]. One study has demonstrated that the mTOR, Sp1, Smad3, and PI3K signaling pathways activate the expression of CTGF in keloid fibroblasts [19]. However, the potential mechanism by which CTGF stimulates ECM production in HSs is still not fully defined.
Based on the previous findings in the literature, we first analyzed aberrant miRNAs expression in HS tissues, and then we focused on the effect of miR-143-3p on cell proliferation and apoptosis in hypertrophic scar fibroblasts (HSFs). Furthermore, the signaling pathway of miR-143-3p-mediated ECM production was also determined in the process of hypertrophic scar formation.
Materials and methods
Tissue samples
Thirteen hypertrophic scar (HS) tissues and paired NS tissues were obtained from 13 different patients, who were admitted to the Third Affiliated Hospital of Xi’an Jiaotong University Medical College. All the protocols were approved by the Ethics Committee of Xi’an Jiaotong University Medical College. Each patient provided written informed consent. The collected skin samples were divided into two portions: one was preserved in liquid nitrogen for the preparation of total RNA and total protein lysates, and the other was used for the isolation and culture of fibroblasts.
Cell culture
Cultures of HSFs and normal skin fibroblasts (NSFs) were established as previously described [20]. Explants were maintained in DMEM supplemented with 10 % heat-inactivated fetal bovine serum and 1 % (w/v) penicillin/streptomycin in a 5 % CO2 humidified atmosphere at 37 °C. Fibroblasts obtained at the third to the fifth passages were used in this study, unless indicated otherwise.
miRNA microarray
Total RNA was extracted from the hypertrophic scars and NS of 13 patients. miRNAs were separated from the total RNA using mirVana miRNA purification columns (Ambion, Austin, TX, USA) for miRCURYTM microarray (Exiqon, Vedbaek, Denmark) analysis according to the manufacturer’s instructions. The miRCURY Hy3™/Hy5™ Power labeling kit (Exiqon, Vedbaek, Denmark) was used to detect the signal of miRNA expression. After hybridization, microarray expression data were acquired and then analyzed using GENEPIX PRO (Molecular Ware, Cambridge, MA, USA). Differences between groups were examined for statistical significance with unpaired Student’s t tests. A p value <0.05 was considered statistically significant.
Quantitative real-time PCR
Total RNA was extracted from both NS tissue and fibroblasts and hypertrophic scars and fibroblasts with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). For mRNA quantification, 500 ng of RNA was used for the synthesis of cDNA with reverse transcriptase using the M-MLV First Strand Kit (Taraka, Dalian, China) according to the manufacturer’s instructions. 1 μL of cDNA was used for real-time PCR with GoTaq qPCR Master Mix (Promega, Madison, WI, USA). For each sample, the relative mRNA level was normalized to β-actin expression. The primers for qPCR are listed in Table 1.
For miRNA quantification, the GoScript Reverse Transcription System Kit (Promega, Madison, WI, USA) was used with the stem loop primer. For each sample, the relative mRNA level was normalized by U6. The specific forward primer of miR-143-3p was as follows: 5′-UGAGAUGAAGCACUGUAGCUC-3′. The forward and reverse primers for U6 were 5′-CTCGCTTCGGCAGCACATATACTA-3′ and 5′-ACGAATTTGCGTGTCATCCTTGCG-3′. Relative expression levels of miRNA or mRNA were analyzed using the Bio-Rad C1000 Thermal Cycler (Bio-Rad, Hercules, CA, USA).
Western blot
Total membranes and nuclear extracts were prepared as described previously, and protein concentrations were determined using the BCA assay (Sangon, Shanghai, China). The dilution of primary antibodies was as follows: Col I 1:200, Col III 1:200, α-SMA 1:350, CTGF, 1:1500, Akt 1:1000, p-Akt 1:1000, mTOR 1:1000, and p-mTOR 1:1000 (Sigma, St. Louis, MO, USA). Each membrane was rinsed three times for 15 min and incubated with the secondary horseradish peroxidase-linked antibodies. β-Actin was used as loading reference for data analysis.
Construction of the expression vector
For the expression plasmid construct, the wild-type CTGF cDNA sequence without the 3′-UTR was selected and cloned into the pcDNA™3.2-DEST vector (Invitrogen, Carlsbad, CA, USA).
Transfection of miR-143-3p mimic and inhibitor
HSFs were seeded 24 h prior to transfection into 24-well or 6-well plates or 6 cm dishes. An miRNA mimic control, miR-143-3p mimic, miRNA inhibitor control, or miR-143-3p inhibitor (Applied Biosystems, Foster City, CA, USA) was transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) at a final concentration of 80 nM. The cells were harvested at 24 h (for RNA extraction), 48 h (for protein extraction), or 72 h (for apoptosis assays).
Cell Counting Kit-8 (CCK-8) assay
Cell proliferation was measured using the CCK-8 kit (Solarbio, Beijing, China). In brief, 48 h after transfection with the miR-143-3p mimic or miR-143-3p inhibitor, HSFs were seeded at 5 × 103 cells per well in 96-well plates in triplicate. At 0, 24, 48, and 72 h, 10 μL of CCK-8 solution mixed with 90 μL of DMEM was added to each well. After 2 h of incubation, absorbance was measured at 450 nm.
Apoptosis assay
HSFs were transfected with 80 nM miR-143-3p mimic or 80 nM miR-143-3p inhibitor, and 48 h after transfection, the cells then were subjected to an apoptosis assay. Apoptosis was detected by Annexin V/PI staining with an apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, 106 treated cells were incubated with Annexin V/PI for 20 min at room temperature. Apoptosis were then analyzed by flow cytometry. The activity of caspases in HSFs culture supernatants was measured using caspase 3 and caspase 9 activity kits (R&D Systems, Minneapolis, MN, USA). The assays were performed according to the manufacturer’s recommendations.
Luciferase reporter assay
The human CTGF 3′-UTR was amplified and cloned into the XbaI site of the pGL3-control vector (Promega, Madison, WI, USA), downstream of the luciferase gene, to generate the plasmid pGL3-CTGF-3′-UTR. As a control, the pGL3-CTGF-3′-UTR-mut plasmids were also constructed using cDNA fragments containing corresponding mutated nucleotides for miR-143-3p. For the luciferase reporter assay, HSFs were co-transfected with the luciferase reporter vectors and control mimic, miR-143-3p mimic, control inhibitor, or miR-143-3p inhibitor, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). A β-actin promoter Renilla luciferase reporter was used for normalization. After 48 h, luciferase activity was analyzed by the Dual-Luciferase Assay System (Promega, Madison, WI, USA), according to the manufacturer’s protocols.
Statistical analysis
All data are expressed as the mean ± SD of results derived from three independent experiments performed in triplicate. Statistical analysis was performed by Student’s t test and ANOVA. A difference was accepted as significant if p < 0.05.
Results
miR-143-3p expression is downregulated and CTGF expression is upregulated in HS tissues and HSFs
To reveal the potential regulating function of miRNAs in the formation and development of HS, the miRNA expression data obtained using miRNA arrays were analyzed (Table 2). The total RNA extracted from HS tissues and the matched NS tissues were hybridized with miRNA microarray chips. 21 miRNAs were significantly differentially expressed: 16 miRNAs were upregulated in HS tissues by more than 2.0-fold, and five miRNAs were found to be downregulated in HS tissues by more than 2.0-fold. TargetScan database was used to screen miRNA candidates targeted CTGF. As a candidate target miRNA of CTGF, miR-143-3p showed significantly decreased expression in HS tissues compared with NS tissues, while both protein and mRNA expression of CTGF were upregulated in HS tissues (Fig. 1a). The qRT-PCR assay showed lower levels of miR-143-3p expression in HSFs compared to NSFs. Both protein and mRNA expression of CTGF were upregulated in HSFs (Fig. 1b). These data suggest that both the downregulation of miR-143-3p and upregulation of CTGF are involved in hypertrophic scar formation.

Expression of miR-143-3p and CTGF in hypertrophic scar tissues and matched normal skin tissues. a The expression of miR-143-3p and CTGF were validated in HS tissues and the matched NS tissues by qPCR and Western blot analysis. b The expression of miR-143-3p and CTGF validated in NSFs and HSFs by qPCR and Western blot analysis. **p < 0.01, versus control group
miR-143-3p inhibits the expression of ECM production-associated protein in HSFs
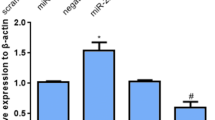
The mRNA and protein expression of Col I, Col III, and α-SMA were detected in HSFs after miR-143-3p or miR-143-3p inhibitor transfection. The expression of miR-143-3p was significantly increased by transfection with miR-143-3p, and significantly reduced by transfection with the miR-143-3p inhibitor (Fig. 2a), suggesting that transfection efficiency was satisfactory for further analysis. The results indicated that the treatment with miR-143-3p reduced the expression of Col I, Col III, and α-SMA mRNA in HSFs. On the contrary, the miR-143-3p inhibitor upregulated the expression of Col I, Col III, and α-SMA mRNA (Fig. 2b). The protein extracted from transfected HSFs assessed by Western blot indicated that Col I, Col III, and α-SMA were downregulated, similarly to the expression of mRNA in transfected HSFs (Fig. 2c). Thus, these findings imply that the miR-143-3p inhibit the expression of ECM production-associated protein in HSFs.

Effects of miR-143-3p on the expression of ECM production-associated proteins in HSFs. a Expression of miR-143-3p was detected by qPCR after miR-143-3p or miR-143-3p inhibitor transfection. b The protein expression of Col I, Col III, and α-SMA in the HSFs was detected by Western blot. c Densitometric analysis of Col I, Col III, and α-SMA protein expression in each group. d qPCR confirmed the mRNA expression of Col I, Col III, and α-SMA in the HSFs. *p < 0.05, versus control group
miR-143-3p inhibits the cell proliferation and promoted apoptosis of HSFs
To further determine the role of miR-143-3p in hypertrophic scarring, the CCK-8 assay was used to measure cell proliferation. It was observed that, 24 h after transfection, the proliferative ability of miR-143-3p-transfected cells was decreased, while that of miR-143-3p inhibitor-transfected cells was increased compared to the control cells (Fig. 3a). The effect of miR-143-3p on cell proliferation was corroborated and mimicked by its effect on the growth rate (Fig. 3b), which is consistent with the results of the CCK-8 assay. Furthermore, flow cytometry analysis showed that both early apoptosis and late apoptosis were upregulated in miR-143-3p-transfected HSFs compared to control cells. Inversely, apoptosis was decreased in miR-143-3p inhibitor-transfected HSFs compared to control cells (Fig. 3c). Moreover, the activity of caspase 3 and caspase 9 was significantly increased in miR-143-3p-transfected HSFs but dramatically reduced in miR-143-3p inhibitor-transfected cells compared to the control cells (Fig. 3d). These data indicated that miR-143-3p inhibits cell proliferation and promotes cell apoptosis in HSFs in vitro.

Effects of miR-143-3p on cell proliferation and apoptosis in HSFs. a Cell proliferation was measured by the CCK-8 assay. b The growth rate of HSFs following miR-143-3p or miR-143-3p inhibitor transfection. c Apoptosis was assessed by flow cytometry. d The activity of caspases 3 and 9 was detected by caspase 3 and caspase 9 activity kits. **p < 0.01 versus control group
miR-143-3p directly targets CTGF expression
The potential targets of miR-143-3p were predicted using computer-aided algorithms in Targetscan. CTGF was identified as a potential miR-143-3p target gene. The putative binding site in the 3′-UTR of CTGF is evolutionally conserved among quite a number of species (Fig. 4a). To verify the binding site, the 3′-UTR of CTGF containing the wild-type or mutated seed-sequence of miR-143-3p was cloned for use in a firefly luciferase reporter assay (Fig. 4b). Compared with the control and miR-143-3p inhibitor, miR-143-3p significantly inhibited the relative luciferase activity when co-transfected with the CTGF-UTR reporter plasmid, but miRNA-mediated inhibitory effects were not observed in the mutant reporter transfected cells (Fig. 4c). Western blot showed that the protein expression of CTGF was decreased in miR-143-3p-transfected HSFs compared with control HSFs (Fig. 4d). These results suggest that miR-143-3p suppresses CTGF expression by directly binding to the 3′-UTR of CTGF.

Identification of CTGF as a target of miR-143-3p. a Putative binding site of miR-143-3p within the 3′-UTR of CTGF. The binding site sequences are highly conserved across different species. b According to the sequence of the miR-143-3p binding site within the 3′-UTR of human CTGF, a luciferase reporter with either the wild-type or mutant sequences of CTGF 3′-UTR was constructed, using the pGL3 vector. c Relative luciferase activity in HSFs was measured after 48 h transfection. d Western blot analysis was used to evaluate the expression of CTGF in HSFs. **p < 0.01 versus control group
CTGF overexpression rescues the miR-143-3p-mediated suppressive effect on ECM production-associated proteins by promoting Akt/mTOR phosphorylation
To evaluate the role of Akt/mTOR signaling in miR-143-3p-mediated CTGF in HSFs, we examined the phosphorylation levels of Akt and mTOR in HSFs that had been transfected with miR-143-3p and exogenous CTGF. miR-143-3p overexpression downregulated CTGF mRNA and protein expression and Akt/mTOR phosphorylation, whereas these effects were restored by eukaryotic expression vector-mediated delivery of CTGF (Fig. 5a, b). Thus, the Akt/mTOR signaling pathway, inhibited by miR-143-3p, was activated by CTGF overexpression. Furthermore, the mRNA and protein expression of Col I, Col III, and α-SMA were downregulated in miR-143-3p-transfected HSFs, but the overexpression of CTGF significantly counteracted this inhibitory effect (Fig. 5c, d). Taken together, these data indicate that overexpression of CTGF abrogates the reduction in miR-143-3p-induced Akt/mTOR phosphorylation, suggesting that CTGF is a functional mediator of the effects of miR-143-3p in HSFs.

Inhibition of CTGF overexpression attenuates the effect of miR-143-3p on ECM production-associated protein. a Western blot analysis was used to evaluate the phosphorylation of Akt and mTOR on HSFs. b Densitometric analysis of Akt and mTOR phosphorylation in each group. c Western blot analysis was used to evaluate the expression of CTGF, Col I, Col III, and α-SMA on HSFs. d Densitometric analysis of CTGF, Col I, Col III, and α-SMA protein expression in each group. *p < 0.05 versus control group. # p < 0.05 versus miR-143-3p group
Discussion
Hypertrophic scarring is characterized by the overgrowth of dense fibrous tissue, which results from deep thermal or traumatic injury to the skin. Aberrant expression of miRNAs has the potential to significantly contribute to the management of cutaneous fibrotic diseases [21]. Twenty-one miRNAs were significantly differentially expressed in hypertrophic scars, as indicated by miRNA arrays: 16 miRNAs were upregulated and 5 miRNAs were downregulated in hypertrophic scar tissues. The altered expression of miR-21, miR-199a-5p, and miR-200b has been reported to be involved in the fibrotic process. In addition, downregulation of miR-4328, miR-143-3p and miR-145-5p has been observed in keloid fibroblasts [22]. Our preliminary data show that miR-143-3p, a candidate target miRNA of CTGF, showed significantly decreased expression in HS tissues compared with NS tissues. As we chose to focus on the miRNAs that are in close association with CTGF, miR-143-3p was selected and analyzed further. Recent studies have indicated that increased miR-143 expression suppresses cell proliferation and promotes apoptosis of human prostate cancer cell line PC-3 [23]. Considering the downregulation of miR-143-3p in HS, miR-143-3p may have a hypertrophic scar-suppressive role in HS development. Thus, we have investigated whether miR-143-3p could induce ECM production-associated protein expression, cell proliferation, and apoptosis of HSFs by targeting CTGF in this study.
Massive collagen synthesis and changes are regarded as the main characteristics of hypertrophic scar formation. In a previous study, the expression of collagen type I and collagen type III in HS tissues and HSFs was found to be significantly higher than in NS tissues and NSFs [24]. Excess deposition of ECM protein by fibroblasts, particularly type I and III collagen is responsible for keloid and HS formation. α-SMA is a marker of myofibroblasts, which is responsible for the deposition of collagen and the pathological formation of HS. In our study, the protein expression of Col I, Col III, and α-SMA protein was reduced in miR-143-3p-transfected HSFs by targeting CTGF, suggesting that miR-143-3p works as an anti-fibrotic factor in HSFs.
miRNAs are involved in proliferation and apoptosis in many cells types [6], although their roles in hypertrophic scarring are only beginning to be investigated. Thus, we investigated the effects of miR-143-3p on HSF proliferation and apoptosis, and the results showed that miR-143-3p inhibited proliferation and promoted apoptosis of HSFs. Moreover, increased expression of CTGF was observed in HS tissues and HSFs in this study. Recombinant CTGF potentially enhances the synthesis of ECM proteins, such as collagen or fibronectin [25]. In addition to a direct fibrogenic effect, CTGF can exacerbate TGF-β-induced fibrosis by activating TGF-β and SMAD signaling through promoting the association of TGF-β with its receptor [26]. miRNAs control target mRNA translation and stability by binding to regulatory sites. Research has found that miR-181c and miR-10a are differentially expressed and target uPA and PAI-1 in HSFs, respectively. Thus, the miR-183 inhibitor and miR-10a could induce the degradation of ECM to reverse the HS [27]. Considering the miR-143-3p downregulation and CTGF upregulation observed in HS, we propose that CTGF is a target gene of miR-143-3p in HSFs. Our findings suggest that miR-143-3p regulates the expression of CTGF at both mRNA and protein levels by directly binding to the 3′-UTR of CTGF. Thus, our results indicate that miR-143-3p mediates the proliferation and apoptosis of HSFs, and downregulates the CTGF expression by targeting CTGF directly in HSFs.
The Akt pathway has essential roles in cell proliferation and apoptosis. A previous study has demonstrated that the Akt/mTOR signaling plays an importance role in the development of fibrosis associated with diabetic nephropathy [28], and the mTOR and PI3K signaling pathways have been demonstrated to activate the expression of CTGF in keloid fibroblasts [19]. Our results indicate that the Akt/mTOR signaling pathway was inhibited by miR-143-3p; however, the overexpression of CTGF significantly counteracted the inhibitory effects of miR-143-3p. Furthermore, the downregulated expression of Col I, Col III, and α-SMA was also rescued by CTGF overexpression. Taken together, these results suggest that miR-143-3p mediates the formation of HS, partly attributable to Akt/mTOR inhibition by targeting CTGF.
Hypertrophic scars is a universal response to traumatic insult, but it is insufficiently studied in tissue regeneration. In human, hypertrophic scars is fibroproliferative disorder of excessive wound healing, which is a programmed process that includes inflammation, proliferation, maturation, and reshaping [13]. Binding of CTGF to fibronectin facilitates adhesion and migration of fibroblasts on fibronectin, which plays an important role in wound healing and tissue repair [29, 30]. Our study indicates miRNA-143-3p overexpression could inhibit hypertrophic scarring by decreasing ECM production-associated protein expression trough targeting CTGF. It is demonstrated that miR-143 inhibits IL-13-induced inflammatory cytokine in nasal epithelial cells [31]. Thus, we suspect that inflammatory process is involved in miR-143-3p-mediated effects on formation of hypertrophic scarring. Inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, can promote fibroblast proliferation and the synthesis of ECM [32]. In the progression of renal fibrosis, TNFα-combined with TGF-β increases CTGF levels and reduces mesangial cells proliferation [33]. The increased level of CTGF mRNA also contributes to the liver regeneration in liver injury rats [34]. Besides, a previous study indicated that miRNA-132 is a critical regulator of skin wound healing, which facilitates the transition from the inflammatory phase to the proliferative phase in epidermal keratinocytes [35]. Moreover, the regulatory role of miRNA-143 in cardiac remodeling has been identified in a previous study [36]. According to these findings, we conclude that miR-143-3p and CTGF play important roles in tissue repair, which may be associated with the inflammatory process. Further experimental verification is needed to demonstrate that.
In conclusion, this study shows that miR-143-3p affects hypertrophic scarring by decreasing ECM production-associated protein expression by targeting CTGF via the Akt/mTOR pathway, as well as by inhibiting proliferation and promoting apoptosis of HSFs. Thus, our study provides evidence to determine that miR-143-3p may be a useful target for the management of hypertrophic scarring.
References
Gauglitz GG, Korting HC, Pavicic T et al (2011) Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med 17:113–125
Sidgwick GP, Iqbal SA, Bayat A (2013) Altered expression of hyaluronan synthase and hyaluronidase mRNA may affect hyaluronic acid distribution in keloid disease compared with normal skin. Exp Dermatol 22:377–379
Meyer LJ, Russell SB, Russell JD et al (2000) Reduced hyaluronan in keloid tissue and cultured keloid fibroblasts. J Invest Dermatol 114:953–959
Syed F, Ahmadi E, Iqbal SA et al (2011) Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: clinical implications for lesional site-directed therapy. Br J Dermatol 164:83–96
Kashiyama K, Mitsutake N, Matsuse M et al (2012) miR-196a downregulation increases the expression of type I and III collagens in keloid fibroblasts. J Invest Dermatol 132:1597–1604
Li P, He QY, Luo CQ (2014) Overexpression of miR-200b inhibits the cell proliferation and promotes apoptosis of human hypertrophic scar fibroblasts in vitro. J Dermatol 41:903–911
He L, He X, Lim LP et al (2007) A microRNA component of the p53 tumour suppressor network. Nature 447:1130–1134
Yi R, O’Carroll D, Pasolli HA et al (2006) Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet 38:356–362
Baek D, Villen J, Shin C et al (2008) The impact of microRNAs on protein output. Nature 455:64–71
Bushati N, Cohen SM (2007) microRNA functions. Annu Rev Cell Dev Biol 23:175–205
Liang P, Lv C, Jiang B et al (2012) MicroRNA profiling in denatured dermis of deep burn patients. Burns 38:534–540
Chau BN, Brenner DA (2011) What goes up must come down: the emerging role of microRNA in fibrosis. Hepatology 53:4–6
Shi-Wen X, Leask A, Abraham D (2008) Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev 19:133–144
Jun JI, Lau LF (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10:945–963
Ivkovic S, Yoon BS, Popoff SN et al (2003) Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130:2779–2791
Duisters RF, Tijsen AJ, Schroen B et al (2009) miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104:170–178 (176 p following 178)
Browne JG, Ho SL, Kane R et al (2011) Connective tissue growth factor is increased in pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci 52:3660–3666
Ponticos M, Holmes AM, Shi-wen X et al (2009) Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 60:2142–2155
Khoo YT, Ong CT, Mukhopadhyay A et al (2006) Upregulation of secretory connective tissue growth factor (CTGF) in keratinocyte-fibroblast coculture contributes to keloid pathogenesis. J Cell Physiol 208:336–343
Arakawa M, Hatamochi A, Takeda K et al (1990) Increased collagen synthesis accompanying elevated m-RNA levels in cultured Werner’s syndrome fibroblasts. J Invest Dermatol 94:187–190
Babalola O, Mamalis A, Lev-Tov H et al (2013) The role of microRNAs in skin fibrosis. Arch Dermatol Res 305:763–776
Li C, Bai Y, Liu H et al (2013) Comparative study of microRNA profiling in keloid fibroblast and annotation of differential expressed microRNAs. Acta Biochim Biophys Sin (Shanghai) 45:692–699
Zhou P, Chen WG, Li XW (2015) MicroRNA-143 acts as a tumor suppressor by targeting hexokinase 2 in human prostate cancer. Am J Cancer Res 5:2056–2063
Zhou R, Zhang Q, Zhang Y et al (2015) Aberrant miR-21 and miR-200b expression and its pro-fibrotic potential in hypertrophic scars. Exp Cell Res 339(2):360–366
Fuchshofer R, Ullmann S, Zeilbeck LF et al (2011) Connective tissue growth factor modulates podocyte actin cytoskeleton and extracellular matrix synthesis and is induced in podocytes upon injury. Histochem Cell Biol 136:301–319
Abreu JG, Ketpura NI, Reversade B et al (2002) Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol 4:599–604
Li C, Zhu HY, Bai WD et al (2015) MiR-10a and miR-181c regulate collagen type I generation in hypertrophic scars by targeting PAI-1 and uPA. FEBS Lett 589:380–389
Lu Q, Zuo WZ, Ji XJ et al (2015) Ethanolic Ginkgo biloba leaf extract prevents renal fibrosis through Akt/mTOR signaling in diabetic nephropathy. Phytomedicine 22:1071–1078
Phanish MK, Winn SK, Dockrell ME (2010) Connective tissue growth factor-(CTGF, CCN2)—a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol 114:e83–e92
Tsang M, Leask A (2015) CCN2 is required for recruitment of Sox2-expressing cells during cutaneous tissue repair. J Cell Commun Signal 9:341–346
Teng Y, Zhang R, Liu C et al (2015) miR-143 inhibits interleukin-13-induced inflammatory cytokine and mucus production in nasal epithelial cells from allergic rhinitis patients by targeting IL13Rα1. Biochem Biophys Res Commun 457:58–64
Wang H, Chen Z, Li XJ et al (2015) Anti-inflammatory cytokine TSG-6 inhibits hypertrophic scar formation in a rabbit ear model. Eur J Pharmacol 751:42–49
Cooker LA, Peterson D, Rambow J et al (2007) TNF-alpha, but not IFN-gamma, regulates CCN2 (CTGF), collagen type I, and proliferation in mesangial cells: possible roles in the progression of renal fibrosis. Am J Physiol Renal Physiol 293:F157–F165
Lee CH, Shah B, Moioli EK et al (2015) CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J Clin Invest 125:3992
Li D, Wang A, Liu X et al (2015) MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J Clin Invest 125:3008–3026
Fernandes T, Barauna VG, Negrao CE et al (2015) Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am J Physiol Heart Circ Physiol 309:H543–H552
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mu, S., Kang, B., Zeng, W. et al. MicroRNA-143-3p inhibits hyperplastic scar formation by targeting connective tissue growth factor CTGF/CCN2 via the Akt/mTOR pathway. Mol Cell Biochem 416, 99–108 (2016). https://doi.org/10.1007/s11010-016-2699-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2699-9