
Abstract
Targeted immunotherapy has become a popular research topic in cancer. The development and metastasis of cervical carcinoma are closely related to epidermal growth factor (EGF) and EGF-1 receptor (EGFR). We successfully constructed a single-chain human anti-EGFR antibody (scFv) and truncated protamine (tP) fusion protein (scFV/tP) expression vector using overlap extension PCR. Enzyme-linked immunosorbent assay and gel shift assay showed that the fusion protein retained the DNA and antigen-binding activity of the original antibody. Using the non-viral scFv/tP vector as a delivery tool, small interfering RNA (siRNA) of the human wings apart-like gene (hWAPL) was effectively transfected into cervical cancer HeLa cells. The hWAPL mRNA expression levels were reduced by 97.23 % in contrast with control cells, and the proliferation capability declined by 66.71 %, indicating significant inhibition. The present results provide a novel strategy for targeted gene therapy and siRNA therapy of EGFR-positive cervical cancers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cervical carcinoma is a common malignant tumor. In China, 0.13 million new cervical carcinoma cases are diagnosed annually. Although its pathogenesis is unclear, it is generally believed that the human papilloma virus (HPV) is the main pathogenic factor in cervical cancer [1]. HPV carcinogenesis is linked with many genes, including the human wings apart-like gene (hWAPL). The hWAPL gene is a homologous sequence of the Drosophila WAPL gene; it was discovered recently and is closely associated with cervical carcinoma and HPV. It has been determined that HPV E6 and E7 can lead to a high level of hWAPL expression [2], which plays a key role during the development of cervical cancer [3].
The development and metastasis of cervical cancer are also associated with the epidermal growth factor (EGF) and its receptors. The EGF-1 receptor (EGFR) is related to proliferation, invasion, metastasis, vascular growth, and apoptosis in cervical cancer cells [4]. Targeted immunotherapy of cancer is a current popular research topic, for example, the United States Food and Drug Administration (US FDA) has approved the commercial EGFR monoclonal antibody cetuximab. Compared to the parent antibody, a single-chain variable fragment (scFv) has small molecules, low immunogenicity, and penetrates some biological barriers easily to arrive at the internal tissues or tumors; it can also be formulated as a carrier-coupled biological toxin or chemotherapeutic drug [5]. However, targeted therapy of cervical cancer with scFv has not been reported. Protamine is a basic protein capable of binding with nucleic acid. Truncated protamine (tP) is a short peptide that contains 22 amino acids, reserving its sequences with abundant basic amino acids, and can bind to nucleic acids [6].
In the present study, a scFv/tP fusion protein with antigen and nucleic acid binding activities was constructed by combining the genes for anti-EGFR scFv and tP. A hWAPL small interfering RNA (siRNA) expression vector was bound with the fusion protein and transfected into cervical carcinoma HeLa cells; the antibody directed the targeted delivery of the siRNA; the inhibitory effects on cervical cancer cell proliferation were observed.
Materials and methods
Reagents and cells
Plasmids pBAD-A, pcDNA3, pEGFP-C3, BL21(DE3)pLysS competent Escherichia coli, and an Ni–NTA purification system were purchased from Invitrogen (Carlsbad, CA). Cervical carcinoma HeLa cells were from ATCC (Rockville, MD). Restriction enzymes and ligase were from Takara (Dalian, China). l-Arabinose was from Promega (Madison, WI). Plasmid mini isolation and gel recovery kits were from Youjing Biotech Co. (Hefei, China). Mouse anti-Myc polyclonal antibody was from Qiagen (Valencia, CA). Goat anti-mouse horseradish peroxidase (HRP)-conjugated IgG and diaminobenzidine (DAB) kit was from Boster Biotech (Wuhan, China). Bicinchoninic acid (BCA) protein assay kit was from Pierce Biotechnology (Rockford, IL). The pcDNA3-siRNA-GFP, pcDNA3-siRNACK-GFP, hWAPL siRNA sequence 5′-CGGACUACCCUUAGCACAAUU-3′, and non-targeting siRNA (control siRNA) 5′-GGCCTAGCGCGTCGAGUAAUU-3′ were prepared by Invitrogen Shanghai (Shanghai, China). The tetrazolium (MTT) kit was from Roche Molecular Biochemicals (Mannheim, Germany).
Construction of scFv/tP expression vector
Based on the EGFR Fab-encoding sequences [7], we designed the following primers for the heavy (VH) and light (VL) chain-coding regions (NcoI andEcoRI restriction sites are underlined):
NcoI
VH: sense (HF) 5′-CGCCATGGCATGCGAGGTGCAGCTGGTG GAG TCTG-3′;
antisense (HR) 5′- GGAAGATCTAGAGGAACCACCAGAGGAGCAC CAGGGTG-3′;
VL: sense (LF) 5′-GGTGGTTCCTCTAGATCTTCCTCCTCTGGTGGC GG TGGCTCGGGCGTGGTGGGGAGCTCGCCCTGACTCAGCC-3′;
EcoRI
antisense (LR) 5′-GGAATTCCAGGACCGCCTCCTGGTAGGACGGT CAGCTTGGTCCC-3′.
According to the tP-encoding sequences from GenBank (GenBank No. AK311987.1), we designed the following primers for tP (EcoRI and HindIII restriction sites are underlined):
tP: sense
EcoRI | HindIII |
5′-AATTCGATGTCGCAGCCAGAGCCGGAGCAGATATTACCGCCAGAGACAAAGAAGTCGCAGACGAAGGAGGCGGAGCTGCCGA-3′;
tP: antisense
EcoRI | HindIII |
5′-AGCTTCGGCAGCTCCGCCTCCTTCGTCTGCGACTTCTTTGTCTCTGGCGGTAATATCTGCTCCGGCTCTGGCTGCGACATCG-3′
The EGFR Fab antibody-encoding fragment was cloned from pcDNA-EGFR as previously described [7]. VH and VL fragments were obtained by PCR using pcDNA-EGFR as the template. The PCR conditions were 94 °C for 5 min for pre-denaturing, followed by 25 cycles of denaturing at 94 °C for 30 s, annealing at 60 °C for 30 s, and polymerization at 72 °C for 1 min, and final extension at 72 °C for 10 min. The scFV fragment was amplified by overlap extension PCR with HF and LR primers under the same PCR conditions, and then subcloned into pBAD-A followed by sequencing for confirmation, yielding pBAD-scFv. The sequence of linker for connecting VH and VL was GGSSRSSSSGGGGSGGGG.
The two complementary oligonucleotides containing the tP-encoding sequences were annealed, and then inserted into pBAD-scFv following restriction enzyme digestion. The recombinant vector was then transformed into E. coli DH5α. Positive colonies were confirmed by restriction enzyme digestion and sequencing, yielding pBAD-scFv/tP.
Expression and identification of scFv/tP fusion protein
pBAD-scFv/tP was transformed into E. coli TOP10F′. A single clone was incubated in Luria–Bertani (LB) broth containing ampicillin at 37 °C overnight. Overnight cultures (20 μL) were added into 2 mL LB broth and cultured at 37 °C. When the absorbance at optical density (OD)600 was 0.8–1.0, l-arabinose was added to a concentration of 0.2 g/L. Bacteria were grown with shaking at 200 rpm on a platform shaker for 20 h at 25 °C. Culture supernatant (10 mL) was centrifuged at 5,000×g. The cells were washed once in ice-cold phosphate-buffered saline (PBS), resuspended in 100 μL PBS, and sonicated. The bacterial lysate was clarified by centrifugation at 10,000×g for 20 min. Aliquots of cell lysates containing 50 μg protein were separated by 12 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose filters. The filters were blocked with Tris-buffered saline/Tween-20 buffer (10 mM Tris–HCl, pH 8.0, 0.15 M NaCl, 0.05 % Tween-20) containing 5 % skimmed milk and incubated with an anti-Myc monoclonal antibody overnight, followed by the addition of HRP-conjugated anti-mouse IgG and DAB visualization of the bands.
Abundant expression and purification of scFv/tP
Overnight cultures (100 μL) were added to 10 mL LB broth to enable abundant expression of scFv/tP. The bacterial lysate supernatant was filtered with a 0.22-μm-diameter filter. The crude proteins were loaded onto a column equilibrated with 15 volumes of binding buffer. The fusion protein was eluted over a 20-column volume imidazole gradient (100–250 mmol/L) with a final step to 500 mmol/L imidazole to strip the column of any remaining protein. Protein eluates were concentrated through a 30,000 MWCO (molecular weight cutoff) membrane (Millipore) by centrifugation at 2,000×g at 4 °C for 30 min. The purity of the sample was analyzed by Coomassie Brilliant Blue-stained SDS-PAGE and Western blot analysis.
Enzyme-linked immunosorbent assay
The antigen-binding activity of the expression products was detected by enzyme-linked immunoassay (ELISA). The recombinant human EGFR protein was diluted to 1 μg/mL to coat a 96-well plate. Diluted protein (50 μL) was added to each well and incubated overnight at 4 °C. The plate was washed with PBS and blocked with 5 % bovine serum albumin (BSA) overnight at 4 °C. The coated plate was washed with PBS five times, and different dilutions of scFv/tP were added to each well (1:100, 1:200, 1:300, 1:400, 1:500, 1:600, 1:700, and 1:800). The blank well contained BSA. The plate was incubated for 2 h. After washing with PBS-Tween 20 six times, 100 μL HRP-conjugated anti-mouse IgG (1:5,000) was added to each well, followed by incubation at 37 °C for 30 min. After washing, 50 μL 3,5,5′-tetramethylbenzidine substrate was added to each well for color development, and the absorbance was read at 450 nm.
Gel shift assay
The DNA-binding activity of scFv/tP was analyzed with a gel mobility shift assay. pEGFP-C3 plasmid DNA (1 μg) was incubated with 1, 2, 4, 8, and 16 μg scFv/tP in 0.2 mol/L NaCl solution for 30 min at room temperature. Then, the DNA-scFv/tP complexes were separated by electrophoresis on 10 % agarose gel followed by ethidium bromide staining.
Transfection ability of scFv/tP
HeLa cells (2 × 105 cells/well) were seeded in a 6-well plate and incubated at 37 °C for 24 h to 70 % confluence. ScFv/tP and pEGFP-C3 were diluted with serum-free RPMI 1640 medium to prepare 16 and 1 μg/mL solutions, respectively. The solutions were left at room temperature for 5 min before they were mixed in a 1:1 ratio and incubated at room temperature for 10 min to form the transfection complex. The original medium was discarded and replaced with the transfection complex. RPMI 1640 medium was used as a control. The cells were incubated for 4 h before the medium was discarded, and fresh RPMI 1640 medium containing 10 % fetal bovine serum (FBS) was added for further incubation at 37 °C in an incubator containing 5 % CO2. After 48-h incubation, fluorescence was observed under a fluorescence microscope. The transfection efficacy was analyzed by flow cytometry (FCM) assay on a BD FACSCalibur (BD, USA).
Cytotoxicity of scFv/tP
The effect of scFv/tP on HeLa cell proliferation was examined by MTT assay. HeLa cells (1.0 × 104 cells/well) were plated in a 96-well plate and incubated for 24 h in RPMI 1640 medium containing 5 % FBS at 37 °C in 5 % CO2. RPMI 1640 medium was used to adjust Lipofectamine 2000, and scFv/tP to the following concentration gradient: 50, 100, 200, 300, and 500 μg/mL. RPMI 1640 medium was used as the negative control. Cells were treated with the various concentrations of Lipofectamine 2000 and scFv/tP for 24 h before being replaced with RPMI 1640 medium containing 10 % FBS for further culturing for 67–72 h. Four hours before the end of the incubation, 20 μL 5 g/L MTT was added to each well. The MTT was removed and replaced with 150 μL dimethyl sulfoxide (DMSO) for further incubation for 30 min at 37 °C until the crystals had dissolved. The OD of each well was measured at a test wavelength of 570 nm using a microculture plate reader (ELX800, BioTek Instruments). Each experimental group included five wells and all experiments were repeated six times. The cell relative growth rate (RGR) was derived using the following equation: RGR = (OD treated well)/(OD control well) × 100 %.
scFv/tP-induced hWAPL siRNA transfection
The experiment was conducted using three groups: pcDNA3.1 (blank control), pcDNA3-siRNACK-GFP (negative plasmid transfection), and pcDNA3-SiRNA-GFP (recombinant plasmid transfection). HeLa cells (2.0 × 105 cells/well) were plated in a 6-well plate and incubated for 24 h in RPMI 1640 medium to 90 % confluence. Opti-MEN (250 μL) containing 4 μg plasmid DNA was mixed with 250 μL Opti-MEN containing 10 μg scfv/tP and incubated at room temperature for 20 min. The transfection complex was added to each well and incubated at 37 °C. Six hours after transfection, the transfection complex replaced with fresh RPMI 1640 medium containing 10 % FBS, and further cultured for 48 h. The cells were harvested for RNA isolation.
hWAPL mRNA detection in transfected cells
Logarithmic growth-phase cells (2 × 106) in three groups mentioned above (pcDNA3.1, pcDNA3-siRNACK-GFP, and pcDNA3-SiRNA-GFP) were harvested. Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA purity was assessed by spectrophotometry (A260/A280 > 1.8); RNA integrity was assessed by denaturing agarose gel electrophoresis and comparison of the intensities of the 28S, 18S, and 5S ribosomal RNA bands. Complementary DNA was synthesized from total RNA, followed by PCR using a reverse transcription-PCR kit (Takara) according to the manufacturer’s instructions. Primers were FOR (forward): 5′-ACAGCGCTGAACTGTGTGCTTC-3′ and REV (reverse): 5′-GCAATGTTCCAAATATTCAATC-3′. The PCR conditions were 94 °C for 5 min for pre-denaturing, followed by 35 cycles of denaturing at 94 °C for 40 s, annealing at 58 °C for 40 s, and polymerization at 72 °C for 40 s, and final extension at 72 °C for 10 min. The PCR products were separated by 1.5 % agarose gel electrophoresis, followed by ethidium bromide staining. The target bands were analyzed densitometrically using VisionWorksLS Analysis software (Upland, CA), and the results were calculated as the ratio of the OD relative to that of β-actin.
Effect of siRNA on cell proliferation
Cells in the three groups described above (pcDNA3.1, pcDNA3-siRNACK-GFP, and pcDNA3-SiRNA-GFP) were trypsinized, and 5 × 103 cells/well were plated in a 96-well plate and incubated for 72 h in RPMI 1640 medium. Four hours before the end of the incubation, 20 μL 5 g/L MTT was added to each well. The MTT was removed and replaced with 150 μL DMSO for further 30-min incubation at 37 °C until the crystals had dissolved. The OD of each well was measured using a microculture plate reader at a test wavelength of 490 nm. Each experimental group included five wells, and all experiments were repeated six times.
Statistical analysis
All analyses were performed using SPSS version 18.0 (SPSS, Chicago, IL). All values are expressed as mean ± SD. One-way analysis of variance was used to determine statistically significant differences among the groups, and the means of two groups were derived with Student’s t test. P < 0.05 was considered statistically significant.
Results
Reconstruction and identification of recombinant expression vector
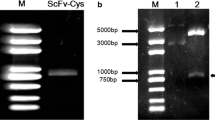
The 405-bp VH gene and 339-bp VL gene were amplified using PCR (Fig. 1a). Using the VH and VL DNA as templates, the 765-bp scFv gene was amplified with overlap extension PCR (Fig. 1b). The PCR product was consistent with the anticipated length and was cloned into the expression vector pBAD-A to construct pBAD-scFv; sequencing confirmed that its sequence was consistent with that of the designed sequence. The artificially synthesized tP gene was then cloned into pBAD-scFv to construct the prokaryotic expression vector pBAD-scFv/tP.

a Heavy (VH), light (VL) and b single-chain genes amplified and assembled by overlap extension PCR. a Lanes M, DNA marker; 1 VH; 2 VL. b Lanes M, DNA marker; 1 single-chain variable fragment
Induced expression and identification of scFv/tP
The expression vector pBAD-scFv/tP was transfected with E. coli TOP10F. The bacterial lysate supernatant was compared with that of control vectors after induction with l-arabinose by 12 % SDS-PAGE. Following induction by 0.2 g l-arabinose, an approximately 36-kD protein band appeared in both the bacterial lysate supernatant and precipitate, which was consistent with the anticipated scFv/tP fusion protein. Western blotting confirmed that the protein was scFv/tP with Myc tags, indicating the successful and soluble expression of the targeted protein (Fig. 2b).

a Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) confirmation of recombinant single-chain human anti-EGFR antibody and truncated protamine fusion protein (scFv/tP) fragment in culture. Lanes M, 0671 protein marker; 1 TOP10F′; 2 scFv/tP supernatant; 3 scFv/tP precipitate. b Western blot of recombinant scFv/tP. Lanes 1, TOP10F′; 2 scFv/tP supernatant; 3 scFv/tP precipitate. c SDS-PAGE of purified recombinant scFv/tP. Lanes M, 0671 protein marker; 1–3, 4, 5, purified scFv/tP eluates from 100, 250, and 500 mM imidazole, respectively. d Western blot of scFv/tP purification. Lanes M, 0671 protein marker; 1, 2, 3, purified scFv/tP eluates from 100, 250, and 500 mM imidazole, respectively
Purification of scFv/tP
SDS-PAGE showed that scFv/tP had been purified effectively using the His-Trap nickel affinity chromatography column technique. SDS-PAGE and Western blotting visualized 36-kD bands, and the protein collected at 250 mmol/L imidazole was purified further (Fig. 2c, d). An average concentration of 3 mg/mL purified protein was tested using the BCA assay.
Antigen-binding characteristics of scFv/tP
ELISA of the recombinant protein showed that the absorbance of 1 mg/mL scFv/tP was 0.86 at 450 nm at a gradient dilution of 1:500; the absorbance was only 0.018 at 450 nm following the BSA reaction. The results suggested that scFv/tP could specifically bind to antigens (Fig. 3).

Enzyme-linked immunosorbent assay of the specific antigen-binding activity of scFv/tP. BSA bovine serum albumin
DNA-binding activity of scFv/tP
The gel shift assay showed that the fusion protein reduced the rate of DNA mobility after 1 μg plasmid DNA was reacted with 1, 2, 4, 8, and 16 μg scFv/tP, with a higher scFv/tP content leading to a slower rate of DNA mobility, indicating that the fusion protein could bind to DNA (Fig. 4a).

a Gel shift assay of scFv/tP binding to plasmid DNA. Lanes 1 1 μg DNA + 16 μg scFv/tP; 2 1 μg DNA + 8 μg scFv/tP; 3 1 μg DNA + 4 μg scFv/tP; 4 1 μg DNA + 2 μg scFv/tP; 5 1 μg DNA + 1 μg scFv/tP; 6: naked DNA. b Fluorescence microscopy of scFv/tP transfection efficiency after 24 h. Lanes 1 control; 2 scFv/tP; 3 green fluorescent control; 4 green fluorescent scFv/tP (×200 magnification). c Flow cytometry (FCM) assay showed the transfection efficiency of scFv/tP was nearly 70 % and exhibited statistical difference to the control group (asterisk)
Transfection capability of scFv/tP
The transfection of scFv/tP was observed under an inverted fluorescence phase-contrast microscope. RPMI 1640 medium was used in the control group (Fig. 4b1–b4). The results of FCM assay indicated that the transfection efficiency of scFv/tP was nearly 70 % and showed statistical difference to the control group (Fig. 4c).
Cytotoxicity of scFv/tP
MTT was added to different concentrations of Lipofectamine 2000 and scFv/tP after HeLa cells had been cultured. The OD value of each group was measured using a microplate reader, and the RGR was calculated (Fig. 5); however, increasing the Lipofectamine 2000 concentration increased the cytotoxicity remarkably.

Cytotoxicity of different doses of scFv/tP and Lipofectamine 2000 in HeLa cells. NC Negative control
Cell transfection with hWAPL mRNA
There was a significant decrease in hWAPL mRNA expression level when cells transfected with hWAPL siRNA were compared with cells in the negative vector-transfected control group and the non-transfected group (Fig. 6a). Analysis with Vision WorksLS Analysis software showed that the band gray value decreased by 96.88 and 97.23 %, respectively, and the difference was statistically significant (P < 0.05). The difference between cells transfected with empty vectors and non-transfected cells was statistically not significant (Fig. 6b).

a Reverse transcription-PCR of hWAPL mRNA expression in HeLa cells. Lanes M, DL 2000 marker; 1 HeLa; 3 pcDNA-siRNACK-GFP; 5 pcDNA-siRNA-GFP; 2, 4, 6, β-actin. b Expression of hWAPL mRNA in HeLa cells. c Effects of hWAPL small interfering RNA on HeLa cell proliferation
Effects of hWAPL siRNA on cell proliferation
The effect of hWAPL downregulation on HeLa cell proliferation was analyzed using the MTT assay. The rate of proliferation of hWAPL siRNA-transfected HeLa cells was decreased by 57.39 and 66.71 % as compared with the negative vector-transfected control cells and non-transfected cells, respectively (P < 0.05) (Fig. 6c).
Discussion
Studies have confirmed that biotherapy with targeted EGFR molecules can suppress the malignant phenotypes of a variety of tumors and can control them effectively, for example, cetuximab and panitumumab are used clinically for several solid cancers [8, 9]. Furthermore, our laboratory and others have successfully used this approach to generate human antibody products for many cancers [10, 11]. However, an antibody can itself be utilized as a drug for immunotherapy as well as be employed as a carrier to selectively deliver a large number of therapeutics to cancer cells by recognition of a specific tumor marker.
As an ideal vector for targeted drugs, scFv has reduced immunogenicity and can enhance antibody specificity and tissue penetrability and include other effector molecules, such as chemotherapeutics, compounds, cytokines, nucleic acids, radionuclides, biotoxins, and other multifunctional molecules constructed by fusion [5, 12, 13]. Although some scFv and their derivatives have presently progressed to clinical Phase I and II trials, scFv targeted therapy in cervical cancer has not been reported yet [14]. Protamine is a novel non-viral natural vector that can bind to DNA, which contains specific nucleic localization signals, and can transport polycation–DNA complexes to the nucleus. It has been approved by the US. FDA for use as a sustained-release agent for insulin and for neutralizing excessive heparin in patients after surgery [15]. In addition, several studies reported the utilization of protamine or scFv/protamine for cancer or HIV therapy [16–19], which made scFv/protamine and siRNA therapy a promising strategy for the treatment of human diseases.
The heavy and light chain variable regions of gene sequences of the human anti-EGFR Fab antibody prepared in our laboratory were connected with the protamine gene using overlap extension PCR to construct the recombinant expression vector of the scFv/tP gene in the present study. After sequencing, the recombinant scFv/tP expression vector was transfected into E. coli TOP10F′. Western blot analysis after l-arabinose induction confirmed the soluble expression of the scFv/tP fusion protein. Via 6-His fusion tags on the vector, high-purity scFv/tP was successfully obtained from the bacterial lysate supernatant using a His-Trap nickel affinity chromatography column. ELISA showed that the fusion protein could bind antigens specifically and had DNA-binding activities. The scFv/tP fusion protein was mixed with plasmid DNA and then transfected into cervical cancer cells. FCM revealed nearly 70 % transfection efficiency of scFv/tP, and no cytotoxicity was detected by MTT analysis.
Discovered recently, the hWAPL gene regulates cell chromosome structure and is homologous to the Drosophila WAPL gene; it is closely associated with cervical cancer and HPV. It is expressed highly and specifically in cervical cancer tissues and possesses cancer gene characteristics. It has been shown that appropriate RNA interference (RNAi) could inhibit hWAPL expression in cells and could promote apoptosis [2]. To further explore the application of scFv/tP as a non-viral vector in tumor therapy, the present study transfected pcDNA-siRNA-GFP into cervical cancer cells through scFv/tP and detected the effects of RNAi on hWAPL expression through reverse transcription-PCR. The hWAPL expression level of cells transfected with pcDNA-siRNA-GFP was reduced by 97.23 % as compared to control cells, indicating the significant inhibition effect of RNAi on hWAPL expression. The effect of hWAPL downregulation on cervical cancer cell growth was observed in vitro via the MTT assay, revealing that the proliferation capacity of siRNA-transfected HeLa cells decreased by 66.71 %, clearly indicating that cell proliferation was inhibited the following hWAPL siRNA transfection using scFv/tP as the vector.
In conclusion, a human anti-EGFR single-chain antibody/tP fusion protein was successfully constructed in this present study. The results suggest that scFv/tP is more effective for treating EGFR-positive cervical carcinoma by helping and facilitating the transfection of hWAPL siRNA. This novel scFv/tP fusion protein provides a promising therapy strategy for cervical carcinoma treatment. In our future research, we will try to optimize the production process, and further investigate the effectiveness in animal models to chart a new path and direction in molecular-targeted therapy for cervical carcinoma.
References
Sundström K, Eloranta S, Sparén P et al (2010) Prospective study of human papillomavirus (HPV) types, HPV persistence, and risk of squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 19:2469–2478
Kuroda M, Kiyono T, Oikawa K et al (2005) The human papillomavirus E6 and E7 inducible oncogene, hWAPL, exhibits potential as a therapeutic target. Br J Cancer 92:290–293
Oikawa K, Akiyoshi A, Tanaka M et al (2008) Expression of various types of alternatively spliced WAPL transcripts in human cervical epithelia. Gene 423:57–62
Soonthornthum T, Arias-Pulido H, Joste N et al (2011) Epidermal growth factor receptor as a biomarker for cervical cancer. Ann Oncol 22:2166–2178
Lin H, Mao Y, Zhang DW et al (2013) Selection and characterization of human anti-MAGE-A1 scFv and immunotoxin. Anticancer Agents Med Chem 13:1259–1266
Inoh Y, Furuno T, Hirashima N et al (2013) Synergistic effect of a biosurfactant and protamine on gene transfection efficiency. Eur J Pharm Sci 49:1–9
Zhang F, Wang X, Tang Q et al (2011) Preparation and characterization of human anti-EGFR scFv–protamine fusion protein. Acta Universitatis Medicinalis Nanjing (Nat Sci) 31:651–655
Nieder C, Pawinski A, Dalhaug A et al (2012) A review of clinical trials of cetuximab combined with radiotherapy for non-small cell lung cancer. Radiat Oncol 7:3
Steele N, Anthony A, Saunders M et al (2012) A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br J Cancer 106:793–798
Chen R, Zhang D, Mao Y et al (2012) A human Fab-based immunoconjugate specific for the LMP1 extracellular domain inhibits nasopharyngeal carcinoma growth in vitro and in vivo. Mol Cancer Ther 11:594–603
Bebb G, Smith C, Rorke S et al (2011) Phase I clinical trial of the anti-EGFR monoclonal antibody nimotuzumab with concurrent external thoracic radiotherapy in Canadian patients diagnosed with stage IIb, III or IV non-small cell lung cancer unsuitable for radical therapy. Cancer Chemother Pharmacol 67:837–845
Kato J, O’Donnell RT, Abuhay M et al (2012) Efficacy and toxicity of a CD22-targeted antibody–saporin conjugate in a xenograft model of non-Hodgkin’s lymphoma. Oncoimmunology 1:1469–1475
Lu D, Zhu Z (2014) Construction and production of an IgG-like tetravalent bispecific antibody, IgG-single-chain Fv fusion. Methods Mol Biol 1060:185–213
Chen ZN, Mi L, Xu J et al (2006) Targeting radioimmunotherapy of hepatocellular carcinoma with iodine (131I) metuximab injection: clinical phase I/II trials. Int J Radiat Oncol Biol Phys 65:435–444
Parodi G, De Luca G, Moschi G et al (2010) Safety of immediate reversal of anticoagulation by protamine to reduce bleeding complications after infarct artery stenting for acute myocardial infarction and adjunctive abciximab therapy. J Thromb Thrombolysis 30:446–451
Ortner A, Wernig K, Kaisler R et al (2010) VPAC receptor mediated tumor cell targeting by protamine based nanoparticles. J Drug Target 18:457–467
Cao M, Deng X, Su S et al (2013) Protamine sulfate-nanodiamond hybrid nanoparticles as a vector for MiR-203 restoration in esophageal carcinoma cells. Nanoscale 5:12120–12125
Hao H, Zhen Y, Wang Z et al (2013) A novel therapeutic drug for colon cancer: EpCAM scFv-truncated protamine (tp)-siRNA. Cell Biol Int 37:860–864
Song E, Zhu P, Lee SK et al (2005) Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 23:709–717
Acknowledgments
This work was supported by grants from the Medical Science and Technique Development Fund, Nanjing, Jiangsu, China (No. ZKX09015), the Nanjing Municipal Science and Technology Commission, Jiangsu, China (No. 20120825), the Medical Science and Technique Development Fund, Nanjing, Jiangsu, China (No. ZKX12025) and the National Natural Science Foundation for Youth of China, China (No. 81301951).
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Huilin Zhang and Yuan Mao contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhang, H., Mao, Y., Zhang, F. et al. The inhibitory effect of a new scFv/tP protein as siRNA delivery system to target hWAPL in cervical carcinoma. Mol Cell Biochem 391, 77–84 (2014). https://doi.org/10.1007/s11010-014-1989-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-1989-3