
Abstract
The present study was aimed to delineate in vivo mechanisms of orally administered fisetin with special reference to mitochondrial dysfunction in lung tissues employing benzo(a)pyrene (B(a)P) as the model lung carcinogen. The recent revival of interest in the study of mitochondria has been stimulated by the evidence that genetic and/or metabolic alterations in this organelle lead to a variety of human diseases including cancer. These alterations could be either causative or contributing factors. Hence, the activities of mitochondrial-specific enzymes of isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, succinate dehydrogenase, malate dehydrogenase and tumor marker, carcinogenic embryonic antigen were analyzed in control and experimental groups of mice. The induction of apoptotic and anti-apoptotic proteins such as Bcl-2/Bax, cytochrome c, caspase-9 and caspase-3 was confirmed by the immunohistochemistry and Western blot analyses. Furthermore, transmission electron microscopy study of lung sections of B(a)P-induced mice showed the presence of phaemorphic cells with dense granules and increased mitochondria. All the aberrations were alleviated when the mice were treated with fisetin (25 mg/kg body weight). The results proved fisetin to be a very successful drug in combating the mitochondrial dysfunction in an experimental model of lung carcinogenesis induced by B(a)P.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is expected to cause 10 million deaths per year worldwide by the year 2030 [1]. The worldwide incidence is 14 %, whereas it constitutes 6.8 % of all cancers in India. It is the leading cancer of both sexes in three of the Urban Cancer Registries (Bhopal, Delhi and Mumbai) in India and is now the leading cause of cancer deaths in women, having surpassed breast cancer [2]. A large number of lung cancers are associated with cigarette smoke, although other factors such as environmental influences and radon or nutrition may also be involved [3]. Tobacco contents of smoke, the polycyclic aromatic hydrocarbons (PAHs) such as benzo(a)pyrene (B(a)P), play a major role in the induction of lung carcinogenesis [4]. B(a)P is metabolized to (±)-B(a)P-r-7,t-8-dihydrodiol-t-9,10-epoxide (BPDE), the ultimate carcinogen. BPDE isomers then bind to the hexocyclic nitrogen of deoxyguanosine in DNA via transaddition of the C-10 position in the epoxide molecule, thus inducing DNA damage, mutation and cancer [5–9].
Mitochondria are vibrant intracellular organelles that generate the supply of adenosine triphosphate (ATP) via oxidative respiration in most of the cells. In addition, they also play an important role in generating apoptotic signals [10]. They are involved either directly or indirectly in many aspects of altered metabolism in cancer cells [11]. For instance, cancer cells are normally addicted to aerobic glycolysis for energy supply since their mitochondrial functions are impaired [12]. Mitochondrial function during apoptosis is controlled by the Bcl-2 family proteins localized to the mitochondrial membranes [13]. The members of the Bcl-2 family of proteins play an important regulatory role. This family consists of the pro- (Bax, Bad,) and anti-apoptotic (Bcl-2, Bcl-xL, etc.) family members. Interactions between these pro- and anti-apoptotic members integrate diverse upstream signals to determine the cellular response, i.e., cytochrome c expulsion from mitochondria [14]. Moreover, the release of cytochrome c is connected with the activation of specific proteases called caspases. In this process, caspase-9 and caspase‐3 seem to play a role in the execution of apoptosis [15]. The increase in caspase‐9 and caspase‐3 activation is synchronized with an alteration of the Bcl‐2 family, such as an increased expression of pro-apoptotic protein Bax and a decreased expression of anti‐apoptotic protein Bcl‐2 [16].
Current scientific interest in the management of cancer is directed toward the utilization of naturally occurring compounds for chemotherapeutics [17]. In recent years, increasing attention has been focused on plant food-derived phytochemicals as potential anticancer drugs. Approximately, 70 % of all drugs used nowadays for the treatment of cancer are derived from natural products [18, 19]. Fisetin (3,7,3,4 tetrahydroxyflavone), one of the major flavonoids widely found in fruits and vegetables [20], is shown to exert antioxidant, anti-inflammatory, anti-angiogenic, anti-invasive and anti-proliferative effects in a wide variety of tumor cells [21–23]. We have recently proved the antioxidant and anticancer potentials of fisetin during experimental lung cancer in mice [24].
The present study is designed to analyze the anticancer potential of orally administered fisetin against the oxidative insult caused by B(a)P in experimentally induced lung carcinogenesis with special emphasis on the defense in the mitochondrial compartment, which is very essential for maintaining the cell viability. At present, there is no information available on the interaction between fisetin and the mitochondrial organelle with reference to Kreb’s cycle enzymes, apoptosis mediators and transmission electron microscopic studies in an in vivo B(a)P-induced lung carcinogenesis model.
Materials and methods
Chemicals
Fisetin and B(a)P were purchased from Sigma chemicals Co (St. Louis, MO, USA). Proteins with primary antibodies, anti-rabbit-Bax, anti-rabbit-Bcl-2 and cytochrome c were procured from Santa Cruz Biotech (USA). Anti-mouse caspase-3 and caspase-9 antibody was generously gifted by Dr. M. Pandi, Madurai Kamaraj University, Madurai, India. All other chemicals and reagents used were of the highest analytical grade commercially available.
Animals
Healthy male Swiss albino mice 6–8 weeks old and weighing 20–25 g were procured from the Tamil Nadu Veterinary and Animal Sciences University (TANUVAS), Madhavaram, Chennai, India. All the experiments were approved by the Institutional Animal Ethical Committee (IAEC), India, guidelines (IAEC No. 04/011/2009). Mice were housed in an air-conditioned room at 22 ± 10 °C with a lighting schedule of 12 h light and 12 h dark. Mice were fed a balanced commercial mice diet (Hindustan UniLever, Mumbai, India) and water ad libitum.
Experimental protocol
The experimental mice were divided into five groups, each group comprising six mice. Group 1 served as normal control. Group 2 mice were administered with B(a)P (50 mg/kg body weight dissolved in corn oil, orally) twice a week for four successive weeks to induce lung cancer by 16th week. Group 3 mice were pre-treated with fisetin (25 mg/kg body weight, dissolved in 0.1 % DMSO, twice a week, orally) 1 week before the first dose of B(a)P induction and continued for 16th weeks. Group 4 mice were post-treated with fisetin (as in group 3) from the 8th week of B(a)P induction till the end of the experiment (16th week). Group 5 mice were treated with fisetin alone for 16 weeks to study the cytotoxicity (if any) induced by fisetin. The dose of fisetin was chosen based on our previous study [24].
Biochemical estimations
Enzyme-linked immunosorbent assay (ELISA) of CEA
Quantitative estimation of the tumor marker carcinoembryonic antigen (CEA) was based on solid phase enzyme-linked immunosorbent assay (ELISA) using the UBI MAGIWELL (USA) enzyme immunoassay kit [25].
Isolation of mitochondria
At the end of the experimental period, mice were anesthetized with diethyl ether followed by cervical decapitation. Lungs and blood were collected; tissues were immediately excised, weighed and then used for the isolation of mitochondria [26]. The activities of tricarboxylic acid (TCA) cycle enzymes, alpha-ketoglutarate dehydrogenase (a-KDH) [27], isocitrate dehydrogenase (ICDH) [28], succinate dehydrogenase (SDH) [29] and malate dehydrogenase (MDH) [30] were assayed.
Immunohistochemical analysis of Bax and Bcl-2
Immunohistochemistry was carried out by the method described in Wiethege et al. [31] on 4 μm paraffin-embedded tissue on poly-l-lysine-coated glass slides. The sections were deparaffinized by placing the slides in an oven at 60 °C for 10 min and then rinsed twice in xylene for 10 min each. The slides were then hydrated in a graded ethanol series (100, 90, 70, 50 and 30 % for 10 min each) and then finally in double-distilled water for 10 min. The sections were incubated with 1 % H2O2 in double-distilled water for 15 min at 22 °C, to quench endogenous peroxidase activity. The sections were rinsed with Tris–HCl containing 150 mM NaCl (pH 7.4) and blocked in a blocking buffer (Tris-buffered saline (TBS), 0.05 % Tween and 3 % BSA for 1 h at 22 °C). After being washed with TBS containing 0.05 % Tween 20, the sections were incubated with primary antibody, rabbit polyclonal IgG to mouse Bax and Bcl-2, at a dilution of 1:1,000 overnight at 4 °C. After incubation, the sections were rinsed with TBS containing 0.05 % Tween 20 twice and incubated with secondary goat anti-rabbit–HRP conjugate, at a dilution of 1:2,000, for 1 h at 4 °C. After another wash with TBS containing 0.05 % Tween 20, the immunoreactivity was developed with 0.05 % 3,3′-diaminobenzidine tetrahydrochloride hydrate (DAB) and 0.01 % H2O2 for 1–3 min. The slides were then counterstained with Meyer’s hematoxylin. To quantify the positive cells, scoring was done as arbitrary units as follows: 4 as intensely stained, 3 as moderately stained, 2 as mildly stained and 1 as poorly stained in control and experimental groups.
Western blot analysis
The whole lungs from each group were homogenized in 10 volumes (v/w) of 20 mM Tris–HCl (pH 7.4) containing 5 mM EDTA and 10 mM mercaptoethanol using a glass–glass Dounce homogenizer. The homogenates were centrifuged at 12,000 rpm, 30 min, 4 °C, and the recovered supernatants were used as lungs protein (50 μg). The lungs proteins prepared from normal and treated mice were electrophoresed by the method described in Laemmli [32] in 12 % SDS-PAGE slab gels. After electrophoretic separation, the proteins were transferred onto Immobilon nitrocellulose membranes. Immunoblotting was performed with primary antibody, that is, Bcl-2, Bax and cytochrome c rabbit polyclonal antibodies (at a dilution of 1:500), caspase-3, caspase-9 and β-actin mouse monoclonal antibody (at a dilution of 1:2,000), after blocking with nonfat dry milk powder and later incubating with peroxidase-tagged anti-rabbit and anti-mouse HRP antibodies, respectively, and the immune complexes were detected using DAB (0.01 %) and H2O2. Quantification of band intensity was done using the Gel Doc system (Bio-Rad) and quantity one software (version 4.0).
Transmission electron microscopy
The changes in the ultrastructure of the lung tissue were studied by electron microscopy according to the method described in An et al. [33]. The lung samples were fixed in Karnovsky’s fixative for 6–8 h at 4 °C. These were post-fixed in 1 % osmium tetraoxide in 0.1 M phosphate buffer for 2 h at 4 °C, dehydrated in ascending grades of acetone, infiltrated and embedded in araldite CY212 and polymerized at 60 °C for 72 h. Thin (60–70 nm) sections were cut with an ultramicrotome. The sections were mounted on copper grids and stained with uranyl acetate and lead citrate and observed under a transmission electron microscope Philip 420.
Statistical analysis
All the grouped data were evaluated with SPSS 10 software. Hypothesis testing methods included one-way analysis of variance (ANOVA) followed by least significant difference test. P values of less than 0.05 were considered to indicate statistical significance. All the results were expressed as mean ± SD for six mice in each group.
Results
Biochemical estimations
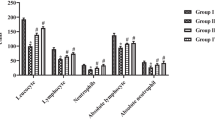
Figure 1 represents the effect of fisetin on the activities of mitochondrial enzymes in the lung of control and experimental mice. The activities of the enzymes, α-KDH, ICDH, SDH and MDH, were found to be significantly (P < 0.05) decreased in carcinogen-treated mice (group 2) when compared to control mice (group 1). On treatment with fisetin (groups 3 and 4), there was a significant increase in the activities of the mitochondrial enzymes when compared with cancer-bearing group. However, there was no significant difference between the control (group 1) and the fisetin-treated mice (group 5).

Effect of fisetin on the activity of mitochondrial enzymes in lung of control and experimental mice. Each value expressed as mean ± SD for six determinations in each experimental groups. Alpha-ketoglutarate dehydrogenase (α-KDH)—mmol of potassium ferrocyanide liberated/min/mg protein; isocitrate dehydrogenase (ICDH)—mmol of α-KG liberated/min/mg protein; succinate dehydrogenase (SDH)—mmol of succinate oxidized/min/mg protein; malate dehydrogenase (MDH)—mmol of NADH oxidized/min/mg protein. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Groups 3 and 4 compared with group 2. c Group 3 compared with group 4
Figure 2 depicts the CEA level of the tumor marker in control and experimental mice. CEA level was found to be significantly increased (P < 0.05) in B(a)P-induced lung cancer-bearing mice (group 2), whereas the level of CEA was significantly decreased upon pre- and post-treatments with fisetin. However, no significant difference was observed between fisetin alone (group 5) and control mice (group 1).

Effect of fisetin on the levels of carcinoembryonic antigen in the serum of control and experimental mice. Each value is expressed as mean ± SD for six mice in each group. CEA levels are expressed as ng/ml. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Group 2 compared with groups 3 and 4. c Group 3 compared with group 4
Immunohistochemical analysis of Bax and Bcl-2
Figures 3 and 4 show the immunohistochemical staining for Bax and Bcl-2, respectively, in the lungs of control and experimental groups of mice. A negligible expression of Bax was observed in B(a)P-induced mice (group 2; Fig. 3b) with subsequent increase in Bcl-2 expression (Fig. 4b), which were significant (P < 0.05) compared to control group (Figs. 3a and 4a). Pre- and post-treatments of fisetin caused a significant (P < 0.05) (groups 3 and 4) increase in the expression of Bax (Fig. 3c, d) and decrease in the expression of Bcl-2 (Fig. 4c, d). Control and fisetin-alone-treated groups (Figs. 3e and 4e) exhibited no significant changes. Quantitative data expressing the corresponding protein levels were assessed using a densitometer that was expressed in relative intensity arbitrary units.

Effect of fisetin on the expression of Bax in control and experimental groups of mice. Plates 1–5 represent the lung section of groups 1–5 of experimental mice, respectively. Rightwards arrow indicates the expression of Bax (values are expressed as mean ± SD for 6 mice each group). Scale bar—100 μm. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Group 2 compared with groups 3 and 4. c Group 3 compared with group 4

Effect of fisetin on the expression of Bcl-2 in control and experimental groups of mice. Plates 1–5 represent the lung section of groups 1–5 of experimental mice, respectively. Rightwards arrow Indicates the expression of Bcl-2 (values are expressed as mean ± SD for 6 mice each group). Scale bar—100 μm. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Group 2 compared with groups 3 and 4. c Group 3 compared with group 4
Western blot analysis
Figure 5 represents the immunoblot analysis of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 in the lungs of control and experimental groups of mice. B(a)P-induced mice (Fig. 5a, Lane 2) showed a decreased expression of Bax with subsequent increase in the level of Bcl-2 as compared to the control mice (Fig. 5a, Lane 1). Pre- and post-treatments with fisetin to B(a)P-induced groups resulted in an increased expression of pro-apoptotic protein Bax (Fig. 5a, Lanes 3 and 4) (P < 0.05), whereas decreases in the level of anti-apoptotic protein Bcl-2. Fisetin-alone-treated group (Fig. 5a, Lane 5) of mice showed a similar pattern as that of the control (Fig. 5a, Lane 1). Quantitative data expressing the corresponding protein level were assessed using a densitometer, and the results were expressed as relative intensity arbitrary units (Fig. 5b).

Immunoblotting and densitometric analyses of Bax and Bcl-2 in the lung of control and experimental groups of mice. a Lanes 1–5 corresponding to the lung tissue lysate of groups 1–5, respectively. b Densitometric analysis of Bax and Bcl-2 immunoblotting. Densitometry data were presented as ‘‘fold change’’ as compared with control. Values are expressed as mean ± SD for 6 mice each group. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Group 2 compared with groups 3 and 4. c Group 3 compared with group 4
Figure 6 shows cytochrome c, caspase-3 and caspase-9 protein expressions in control and experimental groups of mice. There was no significant difference in the expressions between the control (group 1) and fiestin alone group (group 5). On the other hand, there was a significant decrease in the expression of cytochrome c, caspase-3 and caspase-9 in the B(a)P-induced (group 2) animals, whereas, in the fisetin pre- and post-treated (group 3 and group 4) mice, a significant (P < 0.05) increase in caspase-3, caspase-9 and cytochrome c expression (Lanes 3 and 4) was observed, when compared to B(a)P-induced (group 2) lung cancer mice. Quantitative data expressing the corresponding protein level were assessed using a densitometer and the results were expressed as relative intensity arbitrary units (Fig. 6b).

Immunoblotting and densitometric analyses of cytochrome c, caspase-3 and caspase-9 in the lung of control and experimental groups of mice. a Lane 1–5 correspond to the lung tissue lysate of groups 1–5, respectively. b Densitometric analysis of cytochrome c, caspase-3 and caspase-9 immunoblotting. Densitometry data were presented as ‘‘fold change’’ as compared with control. Values are expressed as mean ± SD for 6 mice each group. Statistical significance: P < 0.05. a Group 2 compared with group 1. b Group 2 compared with group 3 and group 4. c Group 3 compared with group 4
Transmission electron microscopy
Figure 7 depicts the consequence of B(a)P and fisetin administrations on the ultrastructural changes in lungs of control and experimental groups of mice. Sections from control group of mice exhibited normal morphology of nucleus, mitochondria and lysosomes (Fig. 7 a–c). B(a)P-induced mouse sections displayed extensive shrunken nucleus, irregular-shaped lysosomes, abnormal changes and complete loss of cristae indicating mitochondrial degeneration (Fig. 7 d–f). Pre- and post-treatments with fisetin to B(a)P-induced mice attenuated the above-mentioned deleterious changes (Fig. 7 g–j). Control mice treated with fisetin also displayed normal morphology of nucleus and mitochondria (Fig. 7 k–l).

Depiction of the consequence of B(a)P and fisetin administrations on the ultrastructural changes in lungs of control and experimental groups of mice. Sections from control group of mice exhibited normal morphology of nucleus, mitochondria and lysosomes (a–c). B(a)P-induced mouse sections displayed extensive shrunken nucleus, showed irregular-shaped lysosomes and abnormal changes and complete loss of cristae indicating mitochondrial degeneration (d–f). Treatment with fisetin to B(a)P-induced mice attenuated the above-mentioned deleterious changes (g–j). Control mice treated with fisetin also displayed normal morphology of nucleus and mitochondria (k and l)
Discussion
The present study is aiming to identify the effect of a plant-based novel bioactive agent in inhibiting experimental lung carcinogenesis. Our results showed that treatment with fisetin could significantly inhibit the growth of lung cancer in mice. In our study, fisetin was able to inhibit the lung cancer by modulating the activities of mitochondrial enzymes and apoptosis. Mitochondria are the energy reservoir of the cell. The damage inflicted to mitochondria would ultimately result in the reduction in energy production, thereby leading to cell death. Subcellular membranes and associated thiol-bearing enzymes represent sensitive sites for carcinogen, causing perturbation of cellular function [34]. Alterations in mitochondrial functions, that is, maintenance of ion homeostasis or ATP supply, have repeatedly been suggested to contribute to cellular transformation [35]. SDH is a marker enzyme in TCA cycle, and succinate, phosphate and ATP promote its activity. Being a regulatory enzyme, its property is altered when it is solubilized. ICDH refers to the NADP+-dependent enzyme, which in several tissues has dual localization being in part cytoplasmic and mitochondrial. Availability of oxalate is controlled by another chief enzyme in the TCA cycle namely MDH, which converts malate to oxaloacetate. In this investigation, decreased activities of major TCA cycle key enzymes such as ICDH, SDH, MDH and α-KDH in lung cancer-bearing mice were observed. Decreased activity of these enzymes might be because of the alteration in cancer cell morphology, ultrastructure, ability of mitochondria to undergo metabolic changes and also drastic reduction in the number of mitochondria in cancer cells [34]. Fisetin supplementation increased the activity of all these TCA cycle enzymes to near-normal levels suggesting its mitochondrion protective nature.
CEA has been extensively studied, particularly in regard to its potential role as a marker of early cancer and prognostic indicator [36]. It is usually over expressed on the cell surface of malignant epithelial-type tumors, and it offers a survival advantage by allowing adhesion into other cells and further metastasis [37]. High CEA levels were also found in 16–23 % of patients with early clinical-stage lung cancer [38, 39]. In this study, an increase in serum CEA levels upon B(a)P treatment was apparently associated with production of tumors. The observed reduction in the levels of CEA in fisetin pre- and post-treated mice was presumably due to decrease in the production rates of tumors that reveal the anti-tumor effect of fisetin. The reduction in the level of CEA was more profound in group 4, fisetin pre-treated mice, than the group 5, fisetin post-treated mice, which shows that fisetin has a more potent anticancer activity when used as a chemopreventive agent.
Apoptosis is an indispensable link in maintaining tissue homeostasis and also an important pathway to eliminate unnecessary cells. Dysregulation of apoptosis is pivotal to tumorigenesis and the development of most cancers. Bcl-2 is a member of anti-apoptotic family of proteins that is found in the outer mitochondrial membrane and can be regulated by post-translational modifications such as phosphorylation. Although the role of phosphorylation of Bcl-2 for its anti-apoptotic function was controversial for a long time, there is growing evidence that phosphorylation regulates Bcl-2 activity [40–42]. Bcl-2 family proteins have also been implicated in the regulation of mitochondrial permeability transition (MPT) pore opening and release of cytochrome c from mitochondria into the cytosol [43], which lead to the activation of caspase-3 and apoptosis [44–46]. Caspases, also called cysteinyl aspartate‐specific proteases, are known for being activated during apoptosis in a self‐amplifying cascade, playing a key role in the execution of apoptosis. Our results revealed that fisetin significantly upregulated the expression of Bax protein and caused simultaneous down-regulation of Bcl-2. Therefore, alterations of Bax/Bcl‐2, cytochrome c and caspase‐3 protein may testify to the apoptosis induced by fisetin in B(a)P-induced lung cancer.
Further results obtained from transmission electron microscopic studies to compare morphological changes between B(a)P-induced and fisetin-treated mice confirmed the chemotherapeutic efficacy of the drug. The cancer-bearing lungs revealed phaemorphic cells, alveolar damages and more number of pyknotic nuclei. Further, the alveolar damage was accompanied by increased number of hyperchromatic, irregular nuclei in the alveolar cells. Moreover, the cytoplasm and nuclei vary in shape. The significant tumor progression observed in the B(a)P-induced mice may be due to the enormous proliferation of the aberrant cancer cells. Fisetin-treated mice showed early and late apoptosis characterized by the presence of irregular and fragmented nuclei, shrunken cells, decreased number of granules in cytoplasm and clumping and alterations in mitochondria. Thus, fisetin plays a key role in the loss of mitochondrial membrane potential leading to apoptosis and inhibition of tumor growth [47].
In conclusion, the results of the present study indicate that fisetin supplementation alleviated mitochondrial dysfunction and induced apoptosis by the upregulation of Bax/Bcl‐2 ratio, thereby leading to cytochrome c release and activation of caspase‐9, caspase‐3 leading to apoptotic cell death during B(a)P-induced lung cancer, thus substantiating its chemopreventive effect.
References
Proctor RN (2001) Tobacco and the global lung cancer epidemic. Nat Rev Cancer 1:82–86
Nandakumar A, Gupta PC, Gangadharan P, Visweswara RN, Parkin DM (2005) Geographic pathology revisited: development of an atlas of cancer in India. Int J Cancer 116:740–754
Ames BN, Gold LS (1998) The causes and prevention of cancer: the role of environment. Biotherapy 11:205–220
Hecht SS, Upadhyaya P, Wang M, Bliss RL, McIntee EJ, Kenney PM (2002) Inhibition of lung tumorigenesis in A/J mice by N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine and myo-inositol, individually and in combination. Carcinogenesis 23:1455–1461
Rojas M, Alexandrov K, Cascorbi I, Brockmöller J, Likhachev A, Pozharisski K, Bouvier G, Auburtin G, Mayer L, Kopp-Schneider A, Roots I, Bartsch H (1998) High benzo[a]pyrene diol-epoxide DNA adduct levels in lung and blood cells from individuals with combined CYP1A1 MspI/Msp-GSTM1*0/*0 genotypes. Pharmacogenetics 8:109–118
Hussain SP, Amstad P, Raja K, Sawyer M, Hofseth L, Shields PG, Hewer A, Phillips DH, Ryberg D, Haugen A, Harris CC (2001) Mutability of p53 hotspot codons to benzo(a)pyrene diol epoxide (BPDE) and the frequency of p53 mutations in nontumorous human lung. Cancer Res 61:6350–6355
Xiong P, Bondy ML, Li D, Shen H, Wang LE, Singletary SE, Spitz MR, Wei Q (2001) Sensitivity to benzo(a)pyrene diol-epoxide associated with risk of breast cancer in young women and modulation by glutathione S-transferase polymorphisms: a case-control study. Cancer Res 61:8465–8469
Alexandrov K, Cascorbi I, Rojas M, Bouvier G, Kriek E, Bartsch H (2002) CYP1A1 and GSTM1 genotypes affect benzo[a]pyrene DNA adducts in smokers’ lung: comparison with aromatic/hydrophobic adduct formation. Carcinogenesis 23:1969–1977
Liang Z, Lippman SM, Kawabe A, Shimada Y, Xu XC (2003) Identification of benzo(a)pyrene diol epoxide-binding DNA fragments using DNA immunoprecipitation technique. Cancer Res 63:1470–1474
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Peluso G, Nicolai R, Reda E, Benatti P, Barbarisi A, Calvani M (2000) Cancer and anticancer therapy-induced modifications on metabolism mediated by carnitine system. J Cell Physiol 182:339–350
Warburg O (1956) On respiratory impairment in cancer cells. Science 124:269–270
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53:4701–4714
Korsmeyer SJ (1992) Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood 80:879–886
Salvesen GS, Dixit VM (1997) Caspases: intracellular signaling by proteolysis. Cell 91:443–446
Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM (1997) Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278:1966–1968
Aggarwal BB, Shishodia S (2006) Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol 71:1397–1421
Newman DJ, Cragg GM, Holbeck S, Sausville EA (2002) Natural products and derivatives as leads to cell cycle pathway targets in cancer chemotherapy. Curr Cancer Drug Targets 2:279–308
Ashokkumar P, Sudhandiran G (2011) Luteolin inhibits cell proliferation during Azoxymethane-induced experimental colon carcinogenesis via Wnt/β-catenin pathway. Invest New Drugs 29:273–284
Arai Y, Watanabe S, Kimira M, Shimoi K, Mochizuki R, Kinae N (2000) Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J Nutr 130:2243–2250
Higa S, Hirano T, Kotani M, Matsumoto M, Fujita A, Suemura M, Kawase I, Tanaka T (2003) Fisetin, a flavonol, inhibits TH2-type cytokine production by activated human basophils. J Allergy Clin Immunol 111:1299–1306
Hanneken A, Lin FF, Johnson J, Maher P (2006) Flavonoids protect human retinal pigment epithelial cells from oxidative-stress-induced death. Invest Ophthalmol Vis Sci 47:3164–3177
Sung B, Pandey MK, Aggarwal BB (2007) Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kappaB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkappaBalpha kinase activation. Mol Pharmacol 71:1703–1714
Ravichandran N, Suresh G, Ramesh B, Siva GV (2011) Fisetin, a novel flavonol attenuates benzo(a)pyrene-induced lung carcinogenesis in Swiss albino mice. Food Chem Toxicol 49:1141–1147
Macnab GM, Urbanowicz JM, Kew MC (1978) Carcinoembryonic antigen in hepatocellular cancer. Br J Cancer 38:51–54
Johnson D, Lardy H (1967) Isolation of liver or kidney mitochondria. Methods Enzymol 10:94–96
Reed LJ, Mukherjee BB (1969) α-Ketoglutarate dehydrogenase complex from Escherichia coli. Methods Enzymol 13:55–61
Slater EC, Borner WD (1952) The effect of fluoride on the succinic oxidase system. Biochem J 52:185–196
Mehler AH, Kornberg A, Grisolia S, Ochoa S (1948) The enzymatic mechanism of oxidation-reductions between malate or isocitrate and pyruvate. J Biol Chem 174:961–977
Birch-Machin MA, Briggs HL, Saborido AA, Bindoff LA, Turnbull DM (1994) An evaluation of the measurement of the activities of complexes I-IV in the respiratory chain of human skeletal muscle mitochondria. Biochem Med Metab Biol 51:35–42
Wiethege T, Voss B, Muller KM (1995) P53 accumulation and proliferating-cell nuclear antigen expression in human lung cancer. J Cancer Res Clin Oncol 121:371–377
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
An Y, Kato K, Nakano M, Otsu H, Okada S, Yamanaka K (2005) Specific induction of oxidative stress in terminal bronchiolar Clara cells during dimethylarsenic-induced lung tumor promoting process in mice. Cancer Lett 230:57–64
Thirunavukkarasu C, Singh JP, Selvendiran K, Sakthisekaran D (2001) Chemopreventive efficacy of selenium against N-nitrosodiethylamine-induced hepatoma in albino rats. Cell Biochem Funct 19:265–271
Pedersen PL (1978) Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res 22:190–274
Veronesi G, Pelosi G, Sonzogni A, Leon ME, D’Aiuto M, Gasparri R, De Braud F, De Pas T, Sandri M, Spaggiari L (2005) Tumour CEA as predictor of better outcome in squamous cell carcinoma of the lung. Lung Cancer 48:233–240
Lin WC, Tseng YT, Chang YL, Lee YC (2007) Pulmonary tumour with high carcinoembryonic antigen titre caused by chronic propolis aspiration. Eur Respir J 30:1227–1230
Sawabata N, Ohta M, Takeda S, Hirano H, Okumura Y, Asada H, Maeda H (2002) Serum carcinoembryonic antigen level in surgically resected clinical stage I patients with non-small cell lung cancer. Ann Thorac Surg 74:174–179
Sawabata N, Hirano H, Inoue M, Okumura Y, Asada H, Takeda S, Maeda H (2003) Immunohistochemical analysis of resected clinical stage I pulmonary adenocarcinomas with high preoperative levels of serum carcinoembryonic antigen. Ann Thorac Surg 76:203–207
Ito T, Deng X, Carr B, May WS (1997) Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem 272:11671–11673
Ruvolo PP, Deng X, May WS (2001) Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 15:515–522
Van Hoof C, Goris J (2003) Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta 1640:97–104
Reed JC, Jurgensmeier JM, Matsuyama S (1998) Bcl-2 family proteins and mitochondria. Biochim Biophys Acta 1366:127–137
Bossy-Wetzel E, Green DR (1999) Apoptosis: checkpoint at the mitochondrial frontier. Mutat Res 434:243–251
Herrera B, Alvarez AM, Sánchez A, Fernández M, Roncero C, Benito M, Fabregat I (2001) Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J 15:741–751
Raha S, Robinson BH (2001) Mitochondria, oxygen free radicals, and apoptosis. Am J Med Genet 106:62–70
Pal HC, Sharma S, Elmets CA, Athar M, Afaq F (2013) Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: role of mitochondrial membrane potential disruption and consequent caspases activation. Exp Dermatol 22:470–475
Acknowledgments
The first author N. Ravichandran gratefully acknowledges University Grants Commission (UGC), for Junior Research Fellowship, New Delhi, India.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ravichandran, N., Suresh, G., Ramesh, B. et al. Fisetin modulates mitochondrial enzymes and apoptotic signals in benzo(a)pyrene-induced lung cancer. Mol Cell Biochem 390, 225–234 (2014). https://doi.org/10.1007/s11010-014-1973-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-1973-y