
Abstract
Red-eared slider turtles, Trachemys scripta elegans, can survive for several weeks without oxygen when submerged in cold water. We hypothesized that anaerobiosis is aided by adaptive up-regulation of the unfolded protein response (UPR), a stress-responsive pathway that is activated by accumulation of unfolded proteins in the endoplasmic reticulum (ER) and functions to restore ER homeostasis. RT-PCR, western immunoblotting and DNA-binding assays were used to quantify the responses and/or activation status of UPR-responsive genes and proteins in turtle tissues after animal exposure to 5 or 20 h of anoxic submergence at 4 °C. The phosphorylation state of protein kinase-like ER kinase (PERK) (a UPR-regulated kinase) and eukaryotic initiation factor 2 (eIF2α) increased by 1.43–2.50 fold in response to anoxia in turtle heart, kidney, and liver. Activation of the PERK-regulated transcription factor, activating transcription factor 4 (ATF4), during anoxia was documented by elevated atf4 transcripts and total ATF4 protein (1.60–2.43 fold), increased nuclear ATF4 content, and increased DNA-binding activity (1.44–2.32 fold). ATF3 and GADD34 (downstream targets of ATF4) also increased by 1.38–3.32 fold in heart and liver under anoxia, and atf3 transcripts were also elevated in heart. Two characteristic chaperones of the UPR, GRP78, and GRP94, also responded positively to anoxia with strong increases in both the transcript and protein levels. The data demonstrate that the UPR is activated in turtle heart, kidney, and liver in response to anoxia, suggesting that this pathway mediates an integrated stress response to protect tissues during oxygen deprivation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mammalian tissues are very sensitive to oxygen limitation and situations of interrupted oxygen supply, such as those occurring during heart attack or stroke, are extremely damaging. In contrast, some ectothermic vertebrates are well-adapted to oxygen limitation. Various species of turtles, for example, have developed capacities for long-term life without oxygen [1, 2]. This includes red-eared sliders (Trachemys scripta elegans) and painted turtles (Chrysemys picta), that can survive without oxygen for several months at cold temperatures [3]. Studies of the anoxia survival mechanisms employed by these animals are not only key to understanding anaerobic lifestyles but can improve our understanding of the mechanisms that prevent hypoxic/ischemic damage in oxygen-sensitive animals. Multiple physiological and metabolic adaptations that support turtle anoxia tolerance have been identified including large tissue stores of fermentable fuels, the ability to both buffer and store lactic acid in the shell, and strong metabolic rate depression that reduces anaerobic metabolic rate by 80–90 % as compared with the corresponding aerobic resting rate at the same temperature [1, 2, 4–6]. Selected gene-regulatory mechanisms are also involved in anoxia tolerance [2, 7], but there is still much to be learned about anoxia-responsive gene expression.
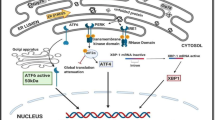
Anoxia is characterized by altered redox conditions and decreased pH, which could disrupt protein folding. Furthermore, anoxia typically quickly triggers an energy-savings plan that includes suppressing most energy-expensive cell functions including ATP-dependent protein synthesis and proteolysis. Hence, there is an increased focus on preserving/protecting existing cellular macromolecules. Previous studies have demonstrated increased production of heat shock proteins and other molecular chaperones in response to anoxia, presumably to help stabilize/protect protein structure/function [8, 9]. Another pathway of potential importance is the unfolded protein response (UPR). The UPR becomes activated in response to the accumulation of unfolded proteins in the endoplasmic reticulum (ER) and consists of several signaling pathways that act to return the ER to its normal physiological state (Fig. 1) [10]. The major actions of the UPR include inhibition of global protein translation to attenuate the influx of new proteins into the ER [11–13], induction of chaperone proteins to increase the protein-folding capacity of the ER [14, 15], and up-regulation of genes involved in the ER-associated degradation pathway to remove proteins that cannot be refolded [16–19].

Overview of the Unfolded Protein Response (UPR). In response to ER stress, the ER-resident stress sensors PERK, IRE1, and ATF6 become activated. Phosphorylated PERK phosphorylates the α subunit of the eukaryotic initiation factor 2 (eIF2α), thereby inhibiting protein synthesis. Phosphorylation of eIF2α also triggers the enhanced expression of activating transcription factor 4 (ATF4), which activates the expression of a variety of target genes
Initiation of the UPR is controlled by three ER-resident protein sensors: two protein kinases, inositol requiring kinase 1 (IRE1) [20, 21] and double-stranded RNA-activated protein kinase-like ER kinase (PERK) [22] and the transcription factor ATF6 (activating transcription factor 6) [21, 23]. PERK is an ER-associated transmembrane serine/threonine protein kinase with a cytoplasmic activity that is selectively activated by ER stress [11, 13]. Upon accumulation of unfolded proteins in the ER lumen, PERK dimerizes and autophosphorylates, leading to its activation [22]. It then phosphorylates a key ribosomal protein, the eukaryotic initiation factor 2 on its alpha subunit (eIF2α) at Ser 51, leading to inactivation of this protein and attenuation of protein synthesis [11, 13]. In addition to the translational attenuation, activation of PERK induces many UPR-dependent genes [11, 24–26]. Its downstream transcription factor, ATF4, is an important stress-responsive transcription factor that is activated upon PERK-mediated phosphorylation of eIF2α [11, 24–26]. ATF4 mediates the transcription of several genes involved in amino acid biosynthesis and transport, anti-oxidant stress responses, and apoptosis [11, 24–26].
Similar to the heat shock response, activation of the UPR could help to relieve cellular stress associated with anoxia and restore cells to homeostasis. In the present study, we explore the responses of multiple UPR-regulated proteins to oxygen deprivation in tissues of the anoxia-tolerant turtle, T. s. elegans. These include ATF4, ATF3, GADD34, GRP78, and GRP94 as well as the phosphorylation state of PERK and eIF2α. The data indicate a tissue-specific activation of the UPR in response to anoxia, suggesting that this pathway mediates an integrated stress response to protect turtle tissues during oxygen deprivation.
Materials and methods
Animals
Adult red-eared slider turtles, T. s. elegans, were purchased from Carolina Biological (Burlington, NC, USA). Turtles had been previously held at 11 ± 1 °C and were initially placed at this temperature in a holding tank containing dechlorinated Ottawa tap water. Temperature was then lowered to 4 ± 1 °C over 1 week and then animals were acclimated at 4 ± 1 °C for 2 weeks before use. Just before experimentation, turtles were moved to 4 °C fridges where they were held for 2 h before experiments were begun. Aerobic control turtles were sampled from this condition; animals were euthanized by decapitation, the shell was opened and then heart, kidney, and liver samples were quickly dissected out (total sampling time 3–5 min) and immediately frozen in liquid nitrogen. To impose anoxia, other turtles were moved into containers filled with dechlorinated water at 4 °C that had been previously bubbled with 100 % nitrogen gas for at least 6 h beforehand. After turtles were placed in a tank, a wire mesh was fitted ~10 cm below the water line so that turtles could not surface during the anoxic submergence; a low level of nitrogen bubbling was continued throughout. Animals were exposed to 5 or 20 h of anoxia and were then sampled as above. All turtles survived the experimental treatments. Animals were cared for in accordance with the guidelines of the Canadian Council on Animal Care and all experimental procedures had the prior approval of the Carleton University Animal Care Committee.
RNA extraction
All solutions and materials were treated with 0.1 % diethylpyrocarbonate (DEPC) and autoclaved before use. Total RNA was isolated from tissues of normoxic control, 5 and 20 h anoxic turtles using Trizol™ (Invitrogen). In brief, 100 mg of tissue was homogenized in 1 mL Trizol using a Polytron homogenizer followed by the addition of 200 μL of chloroform and centrifugation at 10,000×g for 15 min at 4 °C. The upper aqueous phase (containing the RNA) was removed to a fresh tube and precipitated by the addition of 500 μL isopropanol followed by incubation for 10 min at room temperature (RT). The samples were centrifuged again as above and then the RNA pellet was washed with 1 mL of 70 % ethanol followed by a final centrifugation. The resulting pellet was air-dried for 10–15 min and resuspended in 50 μL of DEPC-treated water. The quality of RNA was judged based on the ratio of absorbances at 260 and 280 nm as well as gel electrophoresis on a 1 % agarose gel stained with ethidium bromide to check for the integrity of 18S and 28S ribosomal RNA bands.
Primers
Primers were synthesized by Sigma Genosys. Forward and reverse primers for atf4 and atf3 were designed based on conserved regions of these genes as determined from a DNA alignment of the sequences from other vertebrate species. The forward atf4 primer was 5′-TTCTCCAGCGACCAAGGCTAA-3′ and the reverse was 5′-CCTTGCGGACCTCTTCTATC-3′. Forward and reverse primers for atf3 were 5′-GGTGTCTGCATTGGAAGTC-3′ and 5′-CAAATGCTGCTTCTCATTCT-3′.
The consensus grp78 forward primer was 5′-ACCGCATCACGCCGTCTTAT-3′, whereas the reverse primer was 5′-GCTCCTTGCCATTGAAGAAC-3′. Perfect primers were later designed, the forward being 5′-TTGATGTTGGTGGTGGACAG-3′ and the reverse was 5′-CTTCATGGTGGAACGGAACA-3′. The original grp94 forward primer was 5′-GTGTGGAGCAGCAAGACAGA-3′ and reverse primer was 5′-AGACTGAGTTAAGCGTTGAG-3′. Perfect primers were later designed, the forward being 5′-CGATGATGAGGCTGCTGTTG-3′ and the reverse perfect primer was 5′-CCAGGTTAAGAGTGGCTCAA-3′. The control gene, α-tubulin was amplified with perfect forward (5′-GGAAGATGCTGCCAATAACT’-3′) and reverse (5′-GTCTGGAACTCGGTCAGATC-3′) primers.
cDNA synthesis and PCR
A 4 μg aliquot of RNA was diluted with DEPC water to 10 μL final volume and used to synthesize cDNA. In brief, 1 μL of 200 ng/μL Oligo dT primer was added to the RNA sample and incubated at 65 °C for 5 min. The mixture was then chilled rapidly on ice and 4 μL 5× first strand buffer, 2 μL 100 mM dithiothreitol (DTT), 1 μL dNTP mixture (10 mM each) and 1 μL reverse transcriptase enzyme M-MLV (all reagents from Invitrogen) were added for a total volume 19 μL. The mix was incubated at 42 °C for 1 h and chilled to 4 °C. The resulting cDNA was diluted (10−1–10−3) and amplified by polymerase chain reaction (PCR) with the forward and reverse primers listed above. The PCR reaction of 25 μL final volume was composed by mixing 13.25 μL of sterile water, 5 μL of diluted cDNA, 1.25 μL of primer mixture (final concentration of 1.5 μM forward and 1.5 μM reverse), 2.5 μL of 10× PCR buffer (Invitrogen), 1.5 μL of 50 mM MgCl2, 0.5 μL of dNTP mixture (10 mM each), and 1 μL of Taq Polymerase. The PCR program was as follows: 7 min at 94 °C followed by 36 cycles of 1 min at 94 °C, 1 min at a predetermined annealing temperature (54 °C for atf3 and tubulin, 62.5 °C for atf4), and 1.5 min at 72 °C. The final step was 72 °C for 10 min. PCR products were separated on a 1 % agarose gel stained with ethidium bromide, visualized using the ChemiGenius imaging system (Syngene, Frederick, MD, USA) under UV light and quantified using the GeneTools program. The bands from the most dilute cDNA sample that gave visible product were used for quantification purposes to make sure that the products had not reached amplification saturation.
Amplification of 3′ and 5′ ends
To amplify the 3′ end of atf4, a 4 μg aliquot of RNA was diluted with DEPC water to a 10 μL final volume and used to synthesize cDNA. A 1 μL aliquot of 10 μM AP primer (5′-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3′) was added to the RNA sample and incubated at 65 °C for 5 min. The mixture was then chilled rapidly on ice and 4 μL 5× first strand buffer, 2 μL 100 mM DTT, 1 μL dNTP mixture (10 mM each), and 1 μL reverse transcriptase enzyme M-MLV (all reagents from Invitrogen) were added for a total volume 19 μL. The mix was incubated at 42 °C for 1 h and the reaction was terminated by incubating at 70 °C for 15 min and chilling to 4 °C. The resulting cDNA was diluted 10−1 and then amplified using PCR. The PCR reaction of 25 μL final volume was composed by mixing 12.5 μL of sterile water, 5 μL of diluted cDNA, 1 μL of 10 μM atf4 forward primer, 1 μL of 10 μM AP primer, 2.5 μL of 10× PCR buffer (Invitrogen), 1.5 μL of 50 mM MgCl2, 0.5 μL of dNTP mixture (10 mM each), and 1 μL of Taq Polymerase. The PCR program was as follows: 7 min at 94 °C followed by 36 cycles of 1 min at 94 °C, 1 min at 68 °C, and 1.5 min at 72 °C. The product was ~1,000 bp in size. The 5′ end of atf4 was amplified using First Choice RLM RACE kit (Ambion) using the protocol from the manufacturer. The outer primer used was the atf4 reverse primer, whereas the inner primer was 5′-GCATCCTCCTGGCTGTTATCTGTGTT-3′. The annealing temperature was 62.5 °C. The product obtained was ~500 bp in size.
Sequencing
Polymerase chain reaction products were sequenced by DNA Landmarks (St.-Jean-sur-Richelieu, QC). Sequences were verified as encoding the correct genes using the program BLASTN at the NIH (http://www.ncbi.nlm.nih.gov/blast).
Protein extracts and western blotting
Frozen tissue samples were crushed under liquid nitrogen and then homogenized 1:2.5 w:v in homogenizing buffer (20 mM Hepes, pH 7.5, 200 mM NaCl, 0.1 mM EDTA, 10 mM NaF, 1 mM Na3VO4, 10 mM β-glycerophosphate) with a few crystals of PMSF and 1 μL protease inhibitor cocktail added (Sigma-Aldrich, Oakville, ON, CA). Samples were centrifuged at 4 °C for 15 min at 10,000×g and the supernatant was collected. Soluble protein concentration was measured using the Coomassie Blue dye-binding method with a prepared reagent (Bio-Rad, Hercules, CA, USA) and bovine serum albumin as the standard. Concentrations were adjusted to 10 μg/μL by the further addition of small volumes of homogenizing buffer and then samples were mixed 1:1 v:v with 2× SDS buffer (100 mM Tris-base, 4 % w/v SDS, 20 % v/v glycerol, 0.2 % w/v bromophenol blue, 10 % v/v 2-mercaptoethanol) to give a final protein concentration of 5 μg/μL. Samples were boiled for 5 min and chilled on ice before loading onto gels.
Aliquots of control and anoxic samples containing 30 μg each were loaded onto 10 % polyacrylamide gels, together with prestained molecular weight standards (Bio-Rad) and separated using a discontinuous buffer system [27]. Gels were run in a Mini-Protean III apparatus (Bio-Rad) at a constant voltage (180 V) for 45 min at RT. The resolved proteins were transferred onto PVDF membranes at 70 V for 90 min at 4 °C.
Blots were blocked for 10–30 min in Tris-buffered saline containing 0.05 % Tween-20 (TBST) with 2.5–5 % milk. The blots were then probed overnight with primary antibodies for p-PERK (Thr 981) (Santa Cruz), p-eIF2α (Ser51) (Cell Signaling), ATF4 (Santa Cruz), ATF3 (Santa Cruz), GADD34 (Santa Cruz), GRP78 (StressMarq), or GRP94 (StressGen). Antibodies were diluted 1:250–1:1,000. After incubation, blots were washed several times with TBST and then incubated at RT for 1.5 h with HRP-linked goat anti-rabbit IgG (Cell Signaling, diluted 1:2,000 in TBST). Immunoblots were developed using enhanced chemiluminescence (ECL) reagents.
Preparation of nuclear extracts
Nuclear extracts were prepared using a slight modification of the method described by Dignam et al. [28]. In brief, 0.5 g samples were homogenized using a Dounce homogenizer in 1 mL of homogenization buffer (10 mM Hepes, 10 mM KCl, 10 mM EDTA, pH 7.9). A 10 μL aliquot of 100 mM DTT and 10 μL of Protease Inhibitor Cocktail (Sigma) were added just before homogenization. Samples were centrifuged at 10,000×g for 10 min at 4 °C and the supernatant (cytoplasmic extract) was removed. The pellet was resuspended in 150 μL of extraction buffer (20 mM HEPES, pH 7.9; 400 mM NaCl; 1 mM EDTA; 10 % v/v glycerol) with DTT and Protease Inhibitor Cocktail (1.5 μL) added as above. Tubes containing the samples were put on ice horizontally on a rocking platform for 1 h. Samples were then centrifuged at 10,000×g for 10 min at 4 °C. The supernatant (nuclear extract) was collected. The extracts were quantified and treated as described above to create samples for immunoblotting. The integrity of the nuclei was confirmed by probing immunoblots of cytoplasmic and nuclear fractions with histone H3 antibody (Cell Signaling; diluted 1:1,000 in TBST).
DNA-binding assay
Nuclear extracts were prepared as above, with the exception that following quantification, the soluble protein concentration was adjusted to 8 μg/μL with extraction buffer without further manipulation. Aliquots containing equal amounts of protein from each sample (10–16 μg/well) were then used to assess the amount of binding by ATF4 to its response element. This was done using an ELISA-type assay that used 96-well microplates that were previously coated with the oligonucleotide corresponding to the DNA-binding element that is recognized by ATF4.
The sequence of the biotin-conjugated probe was 5′-Biotin-CTCCCATTTCCATGACGTCATGGTTA-3′, whereas the complementary sequence was 5′-TAACCATGACGTCATGGAAATGGGAG-3′ (ATF4 response element underlined and bold). Both were purchased from Sigma Genosys, diluted to 500 μM using sterile H2O and subsequently mixed 1:1 (v/v) for a total volume of 20 μL. Probes were then placed in a thermocycler for 10 min at 95 °C and the temperature of the block was slowly cooled to RT. A 40 pmol aliquot of double-stranded, biotinylated probe in 50 μL phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) was then added to each well of a streptavidin-coated microplate (VWR). The plate was incubated at RT for 1 h and washed twice with wash buffer (0.1 % Tween-20 in PBS) and once with PBS. Aliquots of 10–16 μg protein were combined with 50 μL of 1× protein-binding buffer composed of 10 mM Hepes, pH 7.9, 50 mM KCl, 0.5 mM EDTA, 3 mM MgCl2, 10 % v:v glycerol, 0.5 mg/mL BSA, 0.05 % NP-40, 1–2 μg salmon sperm DNA (BioShop), 0.5 mM DTT, and NaCl (a variable amount was used based on the volume of nuclear extract added, final concentration was always 40 mM) and added to the wells. Equal amounts of protein for all experimental conditions were used in each assay, and all experimental samples were always run together. Binding buffer, without protein samples, was added to the negative control wells. Plates were incubated at RT with mild agitation for 75 min followed by four washes with wash buffer. A 60 μL aliquot of the ATF4 antibody (the same one used for western blotting), diluted 1:1,000 in PBS was added to the wells and the plate was incubated for 1 h. The plate was washed as above and then 60 μL of anti-rabbit IgG-HRP (BioShop) diluted 1:2,000 in PBS was added and incubated for 1 h. Following this, the plate was washed as above and 60 μL of tetramethylbenzidine (TMB) was added (Bioshop). Once color was developed, the reaction was stopped by the addition of 1 M HCl and optical density was read at 450 nm with a reference wavelength of 655 nm.
Data and statistics
Bands on agarose or SDS-PAGE gels were scanned using a ChemiGenius Bio-Imaging system and densitometric analysis was performed using the associated GeneTools software (Syngene, Frederick, MD). RT-PCR bands for all genes were normalized against bands for α-tubulin amplified from the same cDNA sample. To control for loading in western blots, all blots were stained with Coomassie blue and the band intensity of the immunoreactive material in each lane was standardized against the combined density of a group of Coomassie stained protein bands that were constant between control and experimental conditions and that were not located close to the protein band of interest, as previously described [8, 29, 30]. To standardize ELISA results, the mean OD from the negative controls was subtracted from the OD of each sample. Mean standardized band densities (or OD’s) ± SEM were then calculated for control and anoxic samples. Data were analyzed using one-way analysis of variance and significant differences between the groups were identified using the Student–Newman–Keuls test. Statistical difference was accepted if P < 0.05.
Results
Phosphorylation of PERK and eIF2α
Changes in the relative phosphorylation state of PERK and eIF2α in turtle tissues in response to anoxia were assessed using immunoblotting. The polyclonal anti-p-PERK (Thr 981) antibody cross-reacted with a protein band at ~140 kDa, the expected size of PERK. All tissues examined showed significant increases (P < 0.05) in the phosphorylation state of PERK in response to anoxia (Fig. 2a). Content of p-PERK in heart was unchanged after 5 h of anoxia exposure, but increased to 1.48 ± 0.11 higher than controls in response to 20 h of anoxia, respectively. Relative phosphorylation of p-PERK in kidney increased by 1.43 ± 0.11 fold in response to 5 h of anoxia and further to 1.99 ± 0.05 fold higher than controls after 20 h. Comparable values for liver were 1.64 ± 0.15 fold and 1.96 ± 0.02 fold higher than controls after 5 and 20 h of anoxia, respectively.

Effect of 5 and 20 h of anoxic submergence on the phosphorylation status of PERK and eIF2α in T. s. elegans tissues as determined by Western immunoblotting. a p-PERK (Thr 981) levels, and b p-eIF2α (Ser 51) levels. Representative immunoblots are provided along with histograms showing mean normalized protein levels. Data are mean ± SEM, n = 3–5 independent trials on tissue from different animals. (a) Significantly different from the corresponding control (P < 0.05); (b) significantly different from the 5 h anoxic value (P < 0.05)
The polyclonal anti-p-eIF2α antibody cross-reacted with a protein band of ~40 kDa, the expected size of eIF2α. Figure 2b shows changes in the relative phosphorylation state of eIF2α (Ser 51) in response to anoxia. In both heart and kidney, levels of p-eIF2α were constant after 5 h of anoxia exposure, but increased significantly to 2.50 ± 0.43 and 1.43 ± 0.09 fold higher than controls, respectively, in response to 20 h of anoxia. In liver, p-eIF2α content rose by 1.79 ± 0.11 fold in response to 5 h of anoxia and remained at 1.81 ± 0.27 fold after 20 h.
cDNA cloning of full-length turtle atf4
Using RT-PCR and primers derived from the consensus sequence of atf4 from other vertebrates, a PCR product of 821 bp was retrieved from the total RNA prepared from turtle liver. The product was confirmed as encoding a portion of the atf4 sequence and then 5′ and 3′ RACE were used to extend the sequence and a final assembled sequence of 1,441 bp, encoding a full-length ATF4 sequence of 349 amino acids, was obtained and submitted to GenBank (GenBank ID: JQ731771). Figure 3 shows the first 360 bp of the nucleotide sequence with its three possible open reading frames (ORFs). The atf4 transcript is translated in an unusual manner that depends on the presence of two upstream open reading frames (uORFs) in the 5′ region of its mRNA (described further in the Discussion) [31]. The turtle sequence contained these two uORFs in its 5′ region as well as the third ORF, beginning at nucleotide 284, that codes for the ATF4 protein.

Partial cDNA sequence of atf4 amplified from liver of T. s. elegans and the translated amino acid sequence from three open reading frames. The start codon for each open reading frame (ORF) is highlighted. ORF1 is bolded and boxed. ORF2 is bolded and underlined. ORF3 containing the actual ATF4 reading frame is bolded and shaded. The full 1,441 bp nucleotide sequence encoding a full-length ATF4 sequence of 349 amino acids was submitted to GenBank (accession number JQ731771)
Transcript and protein expression of ATF4 in turtle tissues
Changes in the relative transcript levels of atf4 in response to 5 and 20 h of anoxia exposure were assessed using RT-PCR. Figure 4a shows representative RT-PCR bands of atf4 amplicons with corresponding α-tubulin bands amplified from the same samples, as well as a histogram showing mean levels in three tissues. Both kidney and liver showed significant increases in atf4 transcripts levels in response to 5 h anoxia exposure, rising to 1.50 ± 0.15 and 1.80 ± 0.11 fold higher than control levels (both P < 0.05), respectively. Transcript levels returned to normoxic values by 20 h in both tissues. However, atf4 transcript levels were constant in heart throughout the anoxic episode.

Effect of 5 and 20 h of anoxic submergence on ATF4 expression in T. s. elegans tissues. a Changes in atf4 transcript levels in response to anoxia, as determined by RT-PCR. Representative atf4 amplicons with corresponding α-tubulin bands are shown along with a histogram showing mean normalized transcript levels. Bands for atf4 were normalized against the corresponding α-tubulin band from the same sample. b Changes in ATF4 protein levels in response to anoxia, as determined by immunoblotting. Representative bands are shown along with a histogram with normalized protein levels. Data are mean ± SEM, n = 3–5 independent trials on tissue from different animals. (a) Significantly different from the corresponding control (P < 0.05); (b) significantly different from the 5 h anoxic value (P < 0.05)
Changes in ATF4 protein levels in response to anoxia were assessed via immunoblotting using a polyclonal anti-ATF4 antibody that cross-reacted with a protein band of ~40 kDa, the expected size for ATF4. ATF4 responded differently to anoxia in each tissue (Fig. 4b). In heart, levels were unchanged after 5 h of anoxia but by 20 h were 1.86 ± 0.34 fold (P < 0.05) higher than aerobic control values. In kidney, ATF4 levels had increased by 2.43 ± 0.12 fold after 5 h of anoxia (P < 0.05) and remained elevated at 2.19 ± 0.41 fold above controls after 20 h (P < 0.05). Liver ATF4 levels increased by 1.60 ± 0.08 fold in response to 5 h of anoxia exposure (P < 0.05) but returned to normoxic levels by 20 h.
Nuclear expression and DNA-binding activity of ATF4 in turtle tissues
In order to assess whether increased ATF4 expression corresponded with increased translocation of this transcription factor into the nucleus, ATF4 presence in nuclear extracts was assessed by immunoblotting (Fig. 5a). In heart, ATF4 presence in the nucleus increased significantly by 1.44 ± 0.16 fold after 5 h of anoxia and rose further to 2.05 ± 0.06 fold higher after 20 h. The same pattern was seen in kidney with ATF4 content in nuclear fractions increased by 1.59 ± 0.1 and 1.68 ± 0.16 fold after 5 and 20 h of anoxia, respectively, as compared with aerobic controls. ATF4 levels increased by 1.42 ± 0.04 fold in nuclear fractions from liver of 5 h anoxic turtles but levels returned to control values by 20 h.

Effect of 5 and 20 h of anoxic submergence on ATF4 activation in T. s. elegans tissues. a Changes of ATF4 protein levels in the nuclear fraction of turtle tissues in response to anoxia, as determined by immunoblotting. b Changes in the DNA-binding activity of nuclear ATF4 in response to anoxia, as determined by a transcription factor binding assay. Other information as in Fig. 4
Changes in the DNA-binding activity of nuclear ATF4 in response to anoxia were assessed using a transcription factor binding assay (Fig. 5b). In all cases, DNA-binding activity paralleled the results seen above for immunoblot analysis of ATF4 presence in nuclear fractions. In heart, relative ATF4-binding to DNA in nuclear extracts was 1.67 ± 0.14 fold higher after 5 h of anoxia exposure (although that rise was not statistically significant) and 2.17 ± 0.24 fold higher after 20 h, as compared with controls. Similarly, kidney ATF4 showed 1.52 ± 0.12 and a further 1.94 ± 0.15 fold increases in binding activity after 5 or 20 h of anoxia, respectively. In liver, ATF4-binding activity increased by 2.32 ± 0.46 fold in nuclear extracts from 5 h anoxic turtles but returned to normoxic values after 20 h.
Expression of ATF3 in response to anoxia
Atf3 is a target gene of ATF4. An atf3 PCR product of 333 bp was retrieved from total RNA prepared from turtle liver, confirmed as encoding a portion of the atf3 sequence by BLAST, and submitted to GenBank (GenBank ID: JQ731772). Figure 6a shows changes in the relative transcript levels of atf3 in response to anoxia. Heart atf3 transcripts remained constant in response to 5 h of anoxic submergence, but rose significantly to 2.08 ± 0.1 fold higher than control values after 20 h of anoxia. In the kidney, atf3 transcript levels also remained stable after 5 h of anoxia, but decreased to 54 ± 7 % of the control value by 20 h (P < 0.05). In liver, atf3 levels were also unchanged after 5 h of anoxia, but decreased strongly to 14 ± 1 % of control values by 20 h of anoxia (P < 0.05).

Effect of 5 and 20 h of anoxic submergence on ATF3 expression in T. s. elegans tissues. a Changes in atf3 transcript levels in response to anoxia, as determined by RT-PCR. b Changes in ATF3 protein levels, as determined by immunoblotting. Other information as in Fig. 4
The effects of anoxia exposure on ATF3 protein levels are shown in Fig. 6b. Using a mammalian ATF3 antibody, a band of ~20 kDa was detected, corresponding to the known molecular weight of ATF3. Expression of ATF3 was unchanged in heart in response to 5 h of anoxia, but increased significantly by 1.62 ± 0.10 fold in response to 20 h. Kidney ATF3 was stable after 5 h anoxia, but decreased to 54 ± 10 % of the control after 20 h of anoxia (P < 0.05). In liver, ATF3 increased by 3.32 ± 0.25 fold in response to 5 h of anoxia (P < 0.05), returning to normoxic values by 20 h.
Expression of GADD34 in response to anoxia
Changes in the relative protein expression of GADD34, another downstream target of ATF4, in response to anoxia were assessed using a polyclonal antibody that cross-reacted with a band at ~75 kDa, corresponding to the known molecular weight of GADD34. Figure 7 shows that GADD34 expression remained stable in heart in response to 5 h of anoxia, but increased significantly to 1.38 ± 0.05 fold higher than the control value (P < 0.05) in response to 20 h of anoxia. In liver, GADD34 protein levels increased by 1.79 ± 0.12 fold after 5 h of anoxia but returned to normoxic values after 20 h of anoxia. GADD34 expression did not change significantly in kidney in response to anoxia.

Effect of 5 and 20 h of anoxic submergence the expression of GADD34 in T. s. elegans tissues. a Representative immunoblots b Histogram showing normalized protein levels. Other information as in Fig. 4
GRP78 expression in response to anoxia
A PCR product of 837 bp was retrieved using consensus grp78 primers from total RNA prepared from turtle liver. The product was confirmed as encoding a portion of the grp78 sequence by BLAST and submitted to GenBank (GenBank ID: JQ731773). Relative transcript levels of grp78 were then assessed using RT-PCR and redesigned perfect primers. Figure 8a shows that grp78 transcripts increased significantly in all three tissues after 5 h of anoxia; levels rose by 1.73 ± 0.20, 1.84 ± 0.22, and 2.18 ± 0.09 fold, respectively, in heart, kidney, and liver. However, in all cases, grp78 transcript levels returned to aerobic control values after 20 h of anoxia.

Effect of 5 and 20 h of anoxic submergence on GRP78 expression in T. s. elegans tissues. a Changes in grp78 transcript levels in response to anoxia, as determined by RT-PCR. b Changes in GRP78 protein levels in response to anoxia, as determined by immunoblotting. Other information as in Fig. 4
Relative GRP78 protein levels were assessed using a mammalian GRP78 antibody that cross-reacted with a band at ~78 kDa. In both heart and kidney, levels of GRP78 remained stable after 5 h of anoxia, but increased to 1.53 ± 0.11 and 1.39 ± 0.02 fold higher than controls, respectively, in response to 20 h anoxia exposure. In liver, GRP78 increased significantly by a 2.16 ± 0.21 fold after 5 h of anoxia and remained at 1.80 ± 0.08 fold after 20 h.
GRP94 expression in response to anoxia
A PCR product of 955 bp was retrieved using consensus grp94 primers from total RNA prepared from turtle liver. The product was confirmed as encoding a portion of the grp94 sequence and submitted to GenBank (GenBank ID: JQ731774). Relative transcript levels of grp94, were then assessed using redesigned perfect primers. Figure 9a shows that grp94 transcripts increased significantly by 2.86 ± 0.31 fold in heart compared with controls in response to 5 h of anoxia and remained elevated at 2.10 ± 0.29 fold higher after 20 h. In liver, a strong 4.50 ± 0.42 fold increase was observed after 5 h of anoxia (P < 0.05), but transcripts returned to normoxic values by 20 h. No change in grp94 transcript levels was seen in kidney during anoxia.

Effect of 5 and 20 h of anoxic submergence on GRP94 expression in T. s. elegans tissues. a Changes in grp94 transcript levels in response to anoxia, as determined by RT-PCR. b Changes in GRP94 protein levels in response to anoxia, as determined by immunoblotting. Other information as in Fig. 4
GRP94 protein responses were assessed using an antibody that cross-reacted strongly with a band at ~94 kDa. Figure 9b shows that GRP94 levels remained stable in heart after 5 h of anoxia but increased significantly by 3.16 ± 0.57 fold after 20 h. Liver displayed a 2.36 ± 0.11 fold increase in GRP94 protein content after 5 h of anoxia with levels returning to control levels by 20 h. No change in GRP94 levels were seen in kidney in response to anoxia.
Discussion
A large body of research has shown that perturbation of ER functions can lead to the accumulation of unfolded proteins in the ER lumen, triggering the UPR [10, 32, 33]. Apart from the main metabolic stress of oxygen deprivation, anoxia also causes decreased intracellular pH (due to lactate accumulation) and an altered redox environment [34–36]. These conditions could potentially interfere with the function of the ER and lead to the accumulation of unfolded proteins, triggering the UPR. Previous studies have shown that activation of stress-responsive proteins (e.g., heat shock proteins) and transcription factors occurs in response to anoxia in turtle tissues [8, 29]. The heat shock response involves the proliferation of molecular chaperones and its activation in turtle tissues highlights the importance of stress-responsive pathways to long-term anoxic survival. Activation of the UPR could have similar protective effects during anoxia and studies with mammalian cells have implicated components of the UPR in hypoxia resistance, showing that cells with compromised PERK-eIF2α-ATF4 signaling are more sensitive to hypoxia [37]. Because of its potential importance to anoxia tolerance, we hypothesized that the UPR response would be activated in turtle tissues. The present study shows that various UPR-related proteins are induced in response to anoxic submergence in turtle tissues, suggesting that this response is an important mechanism of cellular protection under anoxia.
Double-stranded RNA-activated PERK is one of three main stress sensors in the ER. Under basal conditions it is found in an inactive state associated with GRP78 (also known as BiP), but ER stress drives GRP78 to dissociate from PERK, allowing PERK to become activated through phosphorylation and oligomerization [38]. Therefore, an increase in the levels of phosphorylated PERK can be used as a marker for the activation of the UPR. The present data show that anoxia exposure led to an increase in PERK phosphorylation in all tissues examined, with relative phosphorylation rising by 1.43–1.99 fold (Fig. 2a). A time-dependent activation of PERK was seen between different tissues. Soft tissues (kidney, liver) showed significantly increased p-PERK content within 5 h of anoxic submergence, whereas p-PERK was elevated in the heart only after 20 h.
The best characterized substrate of PERK is the α subunit of the eukaryotic initiation factor 2, and phosphorylation of this subunit leads to inhibition of translation initiation, thereby reducing the influx of unfolded proteins into the ER. The present study showed a good correlation between the phosphorylation of PERK and eIF2α in heart, kidney, and liver with levels of p-eIF2α increasing by 1.43–2.50 fold in response to anoxia (Fig. 2b).
In addition to the translational inhibition, PERK-mediated UPR activation also leads to induction of the important stress-responsive transcription factor, ATF4. ATF4 is known to respond to a variety of stresses, including oxygen deprivation (hypoxia/anoxia), amino acid limitation, ER stress, and oxidative stress [24, 39]. ATF4 can function as both a transcriptional activator and a repressor and is involved in the induction of genes involved in mitochondrial function, amino acid metabolism, redox chemistry, and protein folding [24].
The atf4 mRNA transcript has uORFs in its 5′ region. These uORFs play a crucial role in the regulation of ATF4 activation in response to eIF2α phosphorylation [31]. The presence of uORFs in the 5′ UTR of atf4 mRNA has been shown in a variety of species, suggesting that this is an evolutionary conserved mechanism of ATF4 regulation [40]. It has been proposed that translation of the atf4 gene is initiated at uORF1 (Fig. 3) and after synthesis of the uORF1-encoded tripeptide, ribosomes continue binding to the atf4 mRNA and reinitiate translation at the downstream uORF2 coding region. In unstressed cells, where eIF2α phosphorylation is low and abundant levels of eIF2-GTP are present, ribosomes scanning downstream from uORF1 rapidly reinitiate translation at uORF2 [31]. Because uORF2 overlaps with the atf4 protein coding region, ATF4 translation is inhibited. However, under stress conditions, increased phosphorylation of eIF2α reduces the amount of eIF2-GTP available and increases the time required for the scanning ribosome to become competent to reinitiate translation [31]. As a result, after translation of uORF1, reinitiation of translation is delayed, resulting in a bypass of the uORF2 initiation codon and the start of translation at the initiation codon for ATF4 proper. Hence, ATF4 protein rapidly accumulates in response to conditions that elevate eIF2α phosphorylation, such as ER stress. In the present study, cloning of the full-length atf4 cDNA from T. s. elegans showed that two uORFs were present in the 5′ region of turtle atf4 mRNA (Fig. 3). Based on these findings, turtle ATF4 appears to be regulated in the same way as in other vertebrates.
ATF4 showed a tissue-specific activation pattern in response to anoxia in T. s. elegans. In heart, atf4 transcript levels did not change but ATF4 protein increased by 1.86 fold after 20 h of anoxic submergence (Fig. 4). This increase correlated with a 2.50 fold increase in p-eIF2α levels and therefore is likely the result of the translational regulation mechanism described above. By contrast, in both kidney and liver, atf4 transcript levels increased significantly in response to 5 h anoxia exposure suggesting the occurrence of an alternative, early mechanism to elevate ATF4 protein levels (especially in 5 h anoxic kidney). This mechanism could be replaced over time when anoxia is prolonged with the p-eIF2α-mediated mechanism of ATF4 synthesis.
Although total ATF4 levels were elevated under anoxia in heart, kidney, and liver, evidence that ATF4-mediated gene transcription is enhanced is also required. Analysis of nuclear localization found increased amounts of ATF4 protein in nuclear extracts after 5 h in all three tissues tested and continued elevation of nuclear ATF4 content after 20 h in heart and kidney (Fig. 5). Furthermore, DNA-binding activity of the transcription factor also increased in parallel and suggests up-regulation of genes under ATF4 control. Overall, these results show that ATF4 is an important regulator of anoxic survival in this vertebrate facultative anaerobe.
In addition to examining the state of ATF4 activation in response to anoxia, the expression of some ATF4 target genes was also examined in turtle tissues. ATF3 is a transcription factor that lies downstream of ATF4 and is involved in the induction of a program of gene expression responsible for cellular remediation, as well as apoptosis [24]. Studies have characterized ATF3 as an adaptive-response gene [41, 42] that is responsive to a wide variety of signals including cytokines, genotoxic agents, or physiological stresses [41]. In the present study, an anoxia-responsive up-regulation of ATF3 was found in heart at both the gene and protein level, probably responding to ATF4 activation of atf3 expression (Fig. 6). The other tissues, however, displayed different results. Kidney showed no change in atf3 transcript or ATF3 protein levels after 5 h of anoxia exposure and, indeed, both transcript and protein levels decreased to about one-half of control values after 20 h of anoxia. The response in liver was anomalous; ATF4 protein levels and activity were elevated after 5 h of anoxic submergence and so was ATF3 protein (a 3.32-increase) and yet atf3 transcript levels did not change. This may suggest a post-transcriptional mechanism for regulating the translation of atf3, possibly under the influence of ATF4. After 20 h anoxia, atf3 transcript levels dropped to just 14 % of control values and ATF3 protein reverted to control levels indicating a probable lack of involvement of ATF3 in the adaptive adjustments of liver for long-term anoxia survival.
Another important ATF4 target is the GADD34 gene, which is known to be upregulated by ATF4 late in the UPR [43]. GADD34 is required for feedback control of the UPR by targeting the type 1 serine/threonine protein phosphatase to eIF2α and dephosphorylating it [43, 44]. Both heart and liver showed elevated levels of GADD34 under anoxia; in heart GADD34 rose by 1.38 fold after 20 h of anoxia and in liver GADD34 increased by 1.79 fold after 5 h of anoxia. Both of these increases correlated with ATF4 activation during anoxia, suggesting ATF4-dependent up-regulation of the gadd34 gene. The increase in GADD34 in heart after 20 h anoxia suggests that the UPR might be subject to inhibition in this tissue at a later time if anoxia was prolonged. Oddly, whereas GADD34 in liver increased in response to 5 h anoxia, no change in the levels of p-eIF2a were evident at this time. This might suggest that GADD34 plays a role other than negative feedback of the UPR in this tissue.
One of the most important functions of the UPR is the up-regulation of molecular chaperones to enhance the folding capacity of the ER. The anoxia-responsive expression of two main ER chaperones was examined: the glucose-regulated proteins GRP78 and GRP94. These are ER-resident chaperones that are up-regulated by multiple branches of the UPR in response to stress, making them a useful marker for UPR onset [45]. GRP78 expression is known to be up-regulated by all three ER sensors of the UPR, whereas GRP94 is mainly regulated by ATF6 and IRE1. Both of these proteins function in refolding protein substrates, and by doing so relieve ER stress and allow a return to homeostasis [45]. Both transcript and protein levels of these chaperones were elevated in T. s. elegans tissues in response to anoxia, with the exception of GRP94 in kidney that was unchanged (Fig. 8, 9). The transcriptional response was strong at 5 h of anoxia exposure with 1.73–4.50 fold increases in grp78 or grp94 transcript levels in tissues. However, the transcriptional response generally abated by 20 h anoxia except for grp94 transcript levels in heart that remained 2.10 fold higher than controls after 20 h. GRP protein levels were significantly elevated by 1.53–3.16 fold after 20 h of anoxia in heart suggesting a role for them in longer term stabilization of ER function under anoxia. Kidney also showed this pattern with grp78 transcript levels elevated after 5 h of anoxia,whereas GRP78 protein was increased after 20 h of anoxia. The time difference observed between the transcript and protein expression of GRP78 in these tissues could potentially be explained by the significant metabolic rate depression that occurs in turtle tissues in response to anoxia. It is possible that while grp78 transcripts accumulate shortly after onset of anoxia (as evident in Fig. 8), it takes a longer time to translate those transcripts into protein, and therefore no increase in protein levels is visible until the later sampling point. However, GRP94 was not anoxia-responsive in kidney. GRP78 is a downstream target of ATF4 but GRP94 is not so these results suggest that anoxia triggers the PERK-ATF4 branch of the UPR in kidney but perhaps not other branches. In the liver both GRPs were elevated by 2.16–2.36 fold after 5 h of anoxia (and GRP78 remained high after 20 h) suggesting a need for GRP chaperone action in helping to stabilize and reorganize ER function as liver transitions into anaerobic metabolism and undergoes strong metabolic rate suppression.
In summary, the present study demonstrates that anoxia exposure activates the UPR in organs of the anoxia-tolerant turtle, T. s. elegans. Various components of the UPR, particularly those regulated under the PERK arm of the UPR including ATF4, its target genes, and GRPs were elevated under anoxia in heart, kidney, and liver. This indicates an important protective role for the UPR in turtle anoxia tolerance. Activation of the UPR early during anoxic submergence could help insure an appropriate folding state for proteins and prolonged protein stability during the transition from aerobic to anoxic states. While the present study predominantly examined the PERK-eIF2α-ATF4 branch of the UPR, it would be of interest for future studies to analyze the other UPR branches, controlled by ATF6 and IRE1, in more detail.
References
Storey KB (1996) Metabolic adaptations supporting anoxia tolerance in reptiles: recent advances. Comp Biochem Physiol B Biochem Mol Biol 113(1):23–35
Storey KB (2007) Anoxia tolerance in turtles: metabolic regulation and gene expression. Comp Biochem Physiol A Mol Integr Physiol 147(2):263–276
Ultsch GR (2006) The ecology of overwintering among turtles: where turtles overwinter and its consequences. Biol Rev Camb Philos Soc 81(3):339–367
Herbert CV, Jackson DC (1985) Temperature effects on the response to prolonged submergence in the turtle Chrysemys picta bellii. II. Metabolic rate, blood acid-base and ionic changes, and cardiovascular function in aerated and anoxic water. Physiol Zool 58:670–681
Hochachka PW, Lutz PL (2001) Mechanism, origin, and evolution of anoxia tolerance in animals. Comp Biochem Physiol B Biochem Mol Biol 130B:435–459
Jackson DC (1968) Metabolic depression and oxygen depletion in the diving turtle. J Appl Physiol 24:503–509
Krivoruchko A, Storey KB (2010) Forever young: mechanisms of natural anoxia tolerance and potential links to longevity. Oxid Med Cell Longev 3(3):186–198
Krivoruchko A, Storey KB (2010) Regulation of the heat shock response under anoxia in the turtle, Trachemys scripta elegans. J Comp Physiol B 180(3):403–414
Prentice HM, Milton SL, Scheurle D, Lutz PL (2004) The upregulation of cognate and inducible heat shock proteins in the anoxic turtle brain. J Cereb Blood Flow Metab 24(7):826–828
Schröder M (2008) Endoplasmic reticulum stress responses. Cell Mol Life Sci 65(6):862–894
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmicreticulum-resident kinase. Nature 397:271–274
Shi Y, An J, Liang J, Hayes S, Sandusky GE, Stramm LE, Yang NN (1999) Characterization of a mutant pancreatic eIF-2a kinase, PEK, and co-localization with somatostatin in islet delta cells. J Biol Chem 274:5723–5730
Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18:7499–7509
Dorner AJ, Wasley LC, Kaufman RJ (1992) Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J 11:1563–1571
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332:462–464
Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2:379–384
Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K (2006) Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol 172:383–393
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249–258
Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K (2003) A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 4:265–271
Sidrauski C, Walter P (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90(6):1031–1039
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5:897–904
Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 20:6755–6767
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Ron D (2002) Translational control in the endoplasmic reticulum stress response. J Clin Invest 110:1381–1388
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11(5):1475–1489
Krivoruchko A, Storey KB (2010) Molecular mechanisms of turtle anoxia tolerance: a role for NF-kappaB. Gene 450(1–2):63–69
Krivoruchko A, Storey KB (2010) Activation of antioxidant defenses in response to freezing in freeze-tolerant painted turtle hatchlings. Biochim Biophys Acta 1800:662–668
Wek RC, Cavener DR (2007) Translational control and the unfolded protein response. Antioxid Redox Signal 9(12):2357–2371
Schröder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Schröder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569(1–2):29–63
Jackson DC, Ultsch GR (2010) Physiology of hibernation under the ice by turtles and frogs. J Exp Zool A Ecol Genet Physiol 313(6):311–327. doi:10.1002/jez.603
Warren DE, Jackson DC (2008) Lactate metabolism in anoxic turtles: an integrative review. J Comp Physiol B 178(2):133–148. doi:10.1007/s00360-007-0212-1
Jackson DC (2004) Acid-base balance during hypoxic hypometabolism: selected vertebrate strategies. Respir Physiol Neurobiol 141(3):273–283. doi:10.1016/j.resp200401009
Fels DR, Koumenis C (2006) The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 5(7):723–728
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529
Ameri K, Hammond EM, Culmsee C, Raida M, Katschinski DM, Wenger RH, Wagner E, Davis RJ, Hai T, Denko N, Harris AL (2007) Induction of activating transcription factor 3 by anoxia is independent of p53 and the hypoxic HIF signalling pathway. Oncogene 26(2):284–289
Vatten KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101(31):11269–11274
Hai T (2006) The ATF transcription factors in cellular adaptive responses. In: Ma J (ed) Gene expression and regulation. Higher Education Press, Beijing, pp 322–333
Lu D, Wolfgang CD, Hai T (2006) Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem 281(15):10473–10481
Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153(5):1011–1022
Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S (2001) Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol 21(20):6841–6850
Lee AS (2001) The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci 26(8):504–510
Acknowledgments
We thank Jan Storey for editorial review of this manuscript. This research was supported by a discovery grant from the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chairs program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krivoruchko, A., Storey, K.B. Activation of the unfolded protein response during anoxia exposure in the turtle Trachemys scripta elegans . Mol Cell Biochem 374, 91–103 (2013). https://doi.org/10.1007/s11010-012-1508-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1508-3