
Abstract
Apigenin is a naturally occurring plant flavone with strong anti-oxidant and anti-inflammatory activity. While the anticancer properties of Apigenin have been extensively studied, little is known about its effects on endothelial dysfunction. We investigated the effects of Apigenin in EAhy926 endothelial cells exposed to TNFα by evaluating the expression of eNOS and MMP-9, two key molecules in endothelial dysfunction. MMP-9 activity was measured by gel zymography. Western blot analysis was performed to analyze eNOS expression and signal transduction. Treatment with Apigenin (50 μM) counteracted the TNFα-induced expression of eNOS and MMP-9 and the TNFα- triggered activation of Akt, p38MAPK and JNK signalling suggesting that multiple signalling pathways are involved in mediating the protective effects of Apigenin on endothelial function. To better understand the molecular mechanisms underlying the protective effects of Apigenin, we used a pharmacological approach with specific inhibitors. The use of an Akt inhibitor mimicked the inhibitory effects of Apigenin on eNOS and MMP-9 expression, suggesting that eNOS and MMP-9 induction by TNFα depends on Akt activation. The TNFα-induced expression of MMP-9 was also affected by the JNK inhibitor SP600125. No effect on eNOS and MMP-9 expression was observed in the presence of the p38MAPK inhibitor SB203580 or the ERK 1/2 inhibitor PD98059. Pretreatment with ‘classic’ (ERα and ERβ) or ‘non classic’ (GPR30) oestrogen receptor (ER) inhibitors (ICI182,780 and PTX, respectively) counteracted the ability of Apigenin to decrease the TNFα-triggered activation of the Akt pathway. Consistently, the use of both ER inhibitors reversed the inhibitory effects of Apigenin on the TNFα-induced expression of eNOS and, to a lesser extent, MMP-9. We can conclude that Apigenin exerts its inhibitory effect on the TNFα-induced expression of eNOS and MMP-9 through the Akt signalling inhibition generated by ER activation. Oestrogen signalling has been implicated in protection from cardiovascular disease. Therefore, having regard to its ability to bind to ERs, Apigenin may be considered an oestrogen-like molecule to potentially be used against the onset and progression of vascular diseases associated with endothelial dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The initiation and the progression of cardiovascular disease are associated with endothelial dysfunction which results in a dysregulated expression of molecules involved in vascular tone, inflammation, and remodelling [1]. Many cardiovascular risk factors such as aging, smoking, inflammation, hyperlipidaemia and hyperglycaemia, contribute to the pathogenesis of endothelial dysfunction through the modulation of TNFα signalling. This inflammatory cytokine plays a pivotal role in vascular disease development by directly promoting atherogenesis, vascular remodelling, inflammation, and oxidative stress [2].
Many studies suggest that a higher intake of polyphenol and flavonoid antioxidant compounds is associated with a decreased risk of cardiovascular disease [3–5]. It is known that the flavone Apigenin, abundantly present in fruits and vegetables, has strong anti-cancer activity. [6]. However considerable evidence suggests that Apigenin may have a protective effect also in other diseases associated with oxidative and inflammatory stress, such as cardiovascular disorders [7]. The protective role of Apigenin in vessels includes mainly anti-thrombotic and anti-inflammatory effects on endothelial and vascular smooth muscle cells, and platelet and leucocyte adhesion. In particular, with reference to the deleterious effects of TNFα on endothelial function, Apigenin inhibits the TNFα-induced up-regulation of cellular adhesion molecules, LOX-1 and COX-2 in cultured human endothelial cells and in vivo, suggesting that Apigenin may interfere with endothelial dysfunction [8–10]. The precise mechanism by which Apigenin improves cell function is unclear. Apigenin shows an estrogenic activity and its effects appear to be mediated through oestrogen receptor (ER) binding-dependent and -independent pathways [11, 12]. In addition to the ‘classic’ receptors ERα and ERβ, a novel G protein-coupled receptor, known as GPR30, has been described as a ‘non classic’ ER and seems to mediate the rapid non genomic oestrogen signalling [13]. Recent evidence has highlighted the regulatory role of ERs for vascular function. ERs have been implicated in protection from cardiovascular disease in women, and accordingly lack of oestrogens is thought to be partly responsible for accelerated development of atherosclerosis in men and postmenopausal women [14, 15].
The aim of this study is to investigate the effects of Apigenin on endothelial cells exposed to TNFα by evaluating the expression of molecules critical for endothelial function such as endothelial nitric oxide synthase (eNOS) and matrix metalloproteinase (MMP)-9. Furthermore, we investigated whether the ‘classic’ and the ‘non classic’ ERs mediate these effects and the signalling pathway involved in the vaso-protective action of Apigenin.
Methods
Cell culture condition and treatments
EAhy926 endothelial cells were cultured in Dulbecco’s modified Eagle’s medium with 10 % foetal calf serum. For treatment experiments, cells were set in serum-free medium (SFM) for 2 h, treated or not with Apigenin (20 and 50 μM, Sigma-Aldrich, A3145) for 1 h and then with TNFα (1 ng/ml, Sigma-Aldrich, T6674). In the experiments with ICI 182,780 (10 μM, Sigma-Aldrich, I4409), Pertussis Toxin (PTX, 4 μg/ml, Sigma-Aldrich, P-7208), SB203580 (20 μM, Santa Cruz, sc-3533), SP600125 (20 μM, BioMol, BML-EI305R), PD98059 (20 μM, Santa Cruz, sc-3532), and Akt inhibitor (40 μM, Calbiochem, 124005), cells were pretreated with inhibitors for 1 h. After 24 h, cells were lysed in RIPA buffer (5 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 % NP-40, 0.5 % deoxycholic acid, 0.1 % SDS) and conditioned media (CMs) were collected, centrifuged to remove cells and debris, and immediately frozen. For intracellular signalling, confluent cells were preincubated for 6 h in SFM and treated or not for short pulses (5–60 min). Cells were lysed in ice-cold lysis buffer (1 % NP-40, 150 mM NaCl, 50 mM Tris HCl pH 8.0, 5 mM EDTA, 10 mM NaF, 10 mM Na4P2O7, 0.4 mM Na3VO4).
Cell viability assay
Cell viability was evaluated with CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer’s instructions. In brief, EAhy926 were treated with different concentrations of Apigenin (20 and 50 μM) and TNFα (1 ng/ml) in SFM. After 24 h, CellTiter 96® Aqueous One Solution Reagent was added directly to culture wells and MTS tetrazolium was bioreduced by cells into a coloured formazan product soluble in the culture medium. The quantity of formazan product formed was measured by a spectrophotometric plate reader at 490 nm and was directly proportional to the number of living cells in culture.
Zymography for MMP-9
Equivalent protein amounts (by BCA methods) from CM were loaded on SDS-acrylamide gel cast with 0.28 % w/v gelatine (type A), run at 6–8 °C in a water-cooled box, rinsed twice for 30 min in 2.5 % Triton X-100, incubated for 16–18 h at 37 °C in 40 mm Tris–HCl, 0.2 m NaCl, 10 mm CaCl2, stained with 0.2 % Coomassie blue in 50 % methanol, 10 % acetic acid, and destained in 50 % methanol, 10 % acetic acid.
Western blotting
Fifty micrograms of proteins from lysates (100 μg for intracellular signalling) were run on reducing SDS-acrylamide gels. The samples were electrotransferred to nitrocellulose, and the membranes were saturated at room temperature for 1 h in TTBS (20 mm Tris–HCl pH 7.5, 500 mm NaCl, 0.01 % Tween 20, 5 % non-fat milk). The membranes were incubated with primary antibody for 16 h in TTBS, then with horseradish peroxidase-secondary antibody (Cell Signaling Technology) for 1 h in TTBS. The bands were visualized by ECL chemiluminescence (Millipore). The membranes were stripped with a denaturing solution (62.5 mM Tris–HCl pH 6.8, 2 % SDS, 100 mM beta-mercaptoethanol) for 20 min at 50 °C, then re-blotted with polyclonal actin. Bands were quantified by optical densitometry (gel analysis system GeneGenius, Syngene, Cambridge, UK). The primary antibodies used were: polyclonal anti-eNOS (Millipore, 07-520, 1:1,000 dilution), polyclonal anti-phospho-p38MAPK (Cell Signaling Technology, 9211S, 1:400 dilution), polyclonal anti-phospho-Akt (Santa Cruz, sc-7985, 1:200), polyclonal anti-phospho-ERK1/2 (Biosource, 44-680, 1:1,000), polyclonal anti-phospho-JNK (Santa Cruz, sc-6254, 1:300), polyclonal anti-actin (Santa Cruz, sc-1615, 1:1,000 dilution).
Statistical evaluation
Each experiment was repeated three to five times. Data shown are mean ± SE. The statistical significance of the results was determined using ANOVA followed by Fisher test. P < 0.05 was considered significant.
Results
Effects of Apigenin on endothelial dysfunction induced by TNFα in EAhy926 endothelial cells
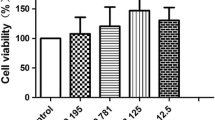
We first performed the MTS test to evaluate cell viability in the presence of Apigenin. As reported in Fig. 1a, Apigenin was not cytotoxic for Eahy926 endothelial cells in the concentrations used in our experiments. Indeed, Apigenin counteracted the decrease of cell viability induced by TNFα treatment. We then examined whether Apigenin modulated the TNFα-induced expression of eNOS and MMP-9, two key molecules associated with endothelial dysfunction. As shown in Fig. 1b, Apigenin decreased in a dose-dependent manner the expression of eNOS induced by TNFα. Similarly, the expression of MMP-9, strictly associated with TNFα treatment, was decreased to basal level by Apigenin (Fig. 1c).

Apigenin reverses the TNFα-induced expression of eNOS and MMP-9 in EAhy926. EAhy926 endothelial cells with or without Apigenin treatment were subjected to TNFα for 24 h. a Cell viability quantified by MTS assay. t0 indicates the viability of cells prior to treatment. Data shown are mean ± SE. b Western blotting of cell lysates and densitometric analysis after actin normalization for eNOS expression. Actin is shown as loading control. c Gelatine zymography of CM and densitometric analysis for MMP-9 activity. Results are shown as fold increase relative to untreated sample. *P < 0.05; **P < 0.01
Effects of Apigenin on TNFα-induced activation of signalling pathways in EAhy926 endothelial cells
Since TNFα stimulation induces the activation of PI3K/Akt, p38MAPK and JNK, we investigated the effects of Apigenin on these signalling pathways. Apigenin pre-treatment (50 μM) significantly inhibited the TNFα-induced phosphorylation of Akt (Fig. 2a), p38MAPK (Fig. 2b) and JNK (Fig 2c). Cells treated with TNFα alone showed a slight but significant induction of ERK1/2 activation, while cells treated with Apigenin showed a further increase in ERK1/2 phosphorylation (Fig. 2d). To determine whether Akt, p38MAPK, and/or JNK could be involved in the induction of eNOS and MMP-9 by TNFα, we used their respective inhibitors [Akt inhibitor, p38MAPK inhibitor (SB203580) and JNK inhibitor (SP600125)] prior to TNFα treatment. Inhibition of Akt by its inhibitor led to a reduction of the TNFα-induced expression of eNOS, while SB203580 and SP600125 had no significant effect (Fig. 2e). Indeed, only a slight effect on MMP-9 expression was detected after treatment with Akt inhibitor and SP600125 (Fig. 2f). Treatment with the ERK1/2 inhibitor did not affect the ability of Apigenin to counteract the induction of eNOS and MMP-9 expression by TNFα (Fig. 2e, f), suggesting that the effect of Apigenin on the TNFα-induced expression of eNOS and MMP-9 did not depend on ERK1/2 phosphorylation. In conclusion, these data show that eNOS induction by TNFα is mediated by Akt activation. The signalling pathway regulating MMP-9 expression is more complex, due to the involvement of Akt and JNK activation.

Apigenin counteracts the TNFα-induced phosphorylation of Akt, p38 MAPK and JNK signalling in EAhy926. EAhy926 endothelial cells with or without Apigenin treatment were subjected to TNFα for 5, 10, 20, 40 and 60 min. Western blotting of cell lysates and densitometric analysis after actin normalization for Akt (a), p38 MAPK (b), JNK (c) and ERK1/2 (d). Results are shown as fold increase relative to t0. Actin is shown as loading control. e Western blotting of cell lysates and densitometric analysis after actin normalization for eNOS expression after treatment with Akt inhibitor, SB203580, SP600125 and PD98059. Actin is shown as loading control. f Gelatine zymography of CM and densitometric analysis for MMP-9 activity after treatment with Akt inhibitor, SB203580, SP600125 and PD98059. Results are shown as fold increase relative to untreated sample. *P < 0.05; **P < 0.01
Involvement of ERs in the inhibitory effect of Apigenin on TNFα-induced eNOS and MMP-9 expression in EAhy926 endothelial cells
It was next investigated whether the ‘classic’ (ERα and ERβ) and/or the ‘non classic’ (GPR30) ERs could mediate the inhibitory effects of Apigenin on the TNFα induction of eNOS and MMP-9 expression. For this purpose, we used a pharmacological approach that included the ERα e ERβ antagonist ICI182,780 and PTX, a potent inhibitor of the G protein-coupled receptor GPR30. As shown in Fig. 3a, the inhibitory effect of Apigenin on the TNFα-induced expression of eNOS was significantly counteracted by ICI182,780 and only partially by PTX treatment. Moreover, the inhibitory effect of Apigenin on the TNFα-induced expression of MMP-9 was slightly reversed by both ICI182,780 and PTX pre-treatment (Fig. 3b). Our data indicate that both ‘classic’ (ERα and ERβ) and ‘non classic’ (GPR30) ERs mediate the inhibitory effect of Apigenin on the TNFα-induced expression of eNOS and MMP-9.

ER inhibitors reverse the inhibitory effect of Apigenin on the TNFα-induced expression of eNOS and MMP-9 in EAhy926. EAhy926 endothelial cells were pre-treated with ICI 182,780 or PTX and then subjected to Apigenin and/or TNFα for 24 h. a Western blotting of cell lysates and densitometric analysis after actin normalization for eNOS expression. Actin is shown as loading control. b Gelatine zymography of CM and densitometric analysis for MMP-9 activity. Results are shown as fold increase relative to untreated sample. *P < 0.05; **P < 0.01
ERs mediate the inhibitory effect of Apigenin on TNFα-induced eNOS and MMP-9 expression through the inactivation of the Akt signalling pathway
To explore whether ERs mediated the inhibitory effect of Apigenin on the TNFα-induced expression of eNOS and MMP-9 through the modulation of the Akt pathway, we evaluated the phosphorylation pattern of Akt after pre-treatment with ER inhibitors ICI182,780 and PTX. As shown in Fig. 4, treatment with ICI182,780 and PTX reversed the effect of Apigenin on the TNFα-triggered activation of Akt. Our results indicate that ERs mediate the inhibitory effects of Apigenin on TNFα-induced eNOS and MMP-9 expression through the inactivation of the Akt signalling pathway.

ER inhibitors reverse the inhibitory effect of Apigenin on the TNFα-induced phosphorylation of Akt. Western blotting and densitometric analysis after actin normalization for Akt phosphorylation after 20 (a) and 40 (b) min in lysates from endothelial cells pre-treated with ICI 182,780 or PTX and then subjected to Apigenin and/or TNFα. Actin is shown as loading control. Results are shown as fold increase relative to sample treated with TNFα. *P < 0.05
Discussion
TNFα is associated with endothelial dysfunction and development of vascular disease. Many recent works have focused on the ability of polyphenols and flavones to improve endothelial function and to influence several markers of cardiovascular risk. Apigenin is a flavone with anti-inflammatory and anti-oxidant properties. It has been suggested that Apigenin may be protective in vascular disorders, although more research needs to be conducted in this regard. For this purpose, in this study we examined the effects of Apigenin on endothelial dysfunction induced by TNFα. The major finding of this study is that Apigenin may interfere with endothelial dysfunction by inhibiting the TNFα-induced expression of eNOS and MMP-9 through a mechanism involving the engagement of both ‘classic’ and ‘non classic’ ERs and the consequent inhibition of Akt signalling.
Some aspects regarding the role of Apigenin in contrasting the deleterious effect of TNFα on endothelial cells still have to be investigated. Other reports showed that Apigenin mitigates the expression of adhesion molecules, COX-2 and LOX-1 induced by TNFα treatment, suggesting a key role of Apigenin in counteracting endothelial dysfunction [8–10]. Our data confirm this hypothesis and indicate that Apigenin counteracts two other key markers of endothelial dysfunction such as MMP-9 and eNOS. MMP-9 is responsible for many complications in vascular diseases, such as vessel wall degradation and angiogenesis [16–18]. Furthermore eNOS is implicated in the regulation of vascular homeostasis and exerts a protective role in the vascular system. However, it has been reported that in the presence of TNFα this positive effect of eNOS on the endothelium is reversed. TNFα selectively transforms eNOS from a nitric oxide (NO)-generating enzyme to an O ·−2 generating enzyme by reducing l-arginine availability to eNOS [19]. TNFα also dramatically increases the level of reactive oxygen species (ROS) by activation of NADPH oxidase activity [20]. The dual activation of the superoxide- and nitric oxide-generating systems provided a favourable environment for protein nitration inducing cytotoxic effects in endothelial cells and connective tissue destruction that are strongly associated with many vascular diseases [21, 22]. Therefore, the induction of eNOS expression after exposure to TNFα represents a futile compensatory mechanism due to a significant decrease in NO bioavailability coupled with dramatic increases in the levels of ROS that further neutralize NO. Therefore, our data indicate that Apigenin plays a protective role in the pathogenesis of TNFα-induced endothelial dysfunction by preventing the deleterious activation of pro-oxidant and proteolytic mechanisms responsible for vascular disease progression.
The precise mechanism by which Apigenin improves endothelial function is unclear. In other cell types, Apigenin effects appear to be mediated through ER binding-dependent and -independent mechanisms involving different signalling pathways [11, 12, 23, 24].
We here report that pre-treatment with Apigenin significantly inhibited the TNFα-triggered activation of Akt, p38MAPK and JNK in endothelial cells. The ability of Apigenin to modulate the activation of Akt, p38MAPK and JNK has already been reported in cancer cells expressing constitutively activated forms of these molecules or after induction by various growth factors [10, 25–27].
Moreover, using a pharmacological approach that included ER inhibitors ICI182,780 and PTX, we here demonstrate the involvement of ERs in mediating the inhibitory effects of Apigenin on the TNFα-induced expression of eNOS and MMP-9. In particular, the expression of eNOS is strongly regulated by Apigenin through the engagement of the ‘classic’ ERs and, to a lesser extent, of a G protein-coupled receptor, presumably GPR30. The experiments with ER inhibitors also indicate that the activation of ERs by Apigenin leads to a rapid non-genomic response which provides for an efficient inhibition of the Akt signalling pathway activated by TNFα. It is known that Akt is one of the pathways involved in the activation of eNOS by TNFα through the involvement of neutral Sphingomyelinase2 [28]. Accordingly, our data indicate that eNOS induction by TNFα depends on Akt activation. Therefore, the ability of Apigenin to inhibit Akt activation through ER engagement leads to the reduction of the TNFα-induced expression of eNOS in EAhy926 cells. Conversely, our data indicate that p38MAPK and JNK inactivation by Apigenin is not involved in eNOS regulation. Interestingly, SB203580 inhibitor increases eNOS expression. This increase may be explained by the ability of SB203580 to enhance eNOS promoter activity [29]. Further experiments should be performed to study extensively the role of p38MAPK activation in eNOS expression. However, we can argue that the maintenance of p-38MAPK activation beyond a threshold level is necessary for the expression of a physiological and protective level of eNOS. If p38MAPK phosphorylation falls below this level, such as after treatment with SB203580, the expression of eNOS may increase dangerously in a pro-oxidant environment. Apigenin probably lowers p38MAPK phosphorylation induced by TNFα to a level that has no effect on eNOS expression. Consistently, our data indicate that in our model the TNFα-induced expression of eNOS depends solely on Akt phosphorylation.
Further studies should be conducted to fully elucidate the molecular mechanisms underlying the inhibitory effects of Apigenin on the induction of MMP-9 expression by TNFα. Our data suggest that these effects are mediated by the modulation of multiple signalling pathways. As indicated by the experiments with inhibitors, the engagement of ERs and the consequent inactivation of Akt signalling by Apigenin mediate only in part the decrease of the TNFα-induced expression of MMP-9. Clearly, this mechanism is not exclusive. Our results indicate that the modulation of MMP-9 expression by Apigenin partially depends also on JNK signalling. Such evidence leads us to hypothesize that Apigenin may use receptors other than ERs and different signalling pathways, as confirmed in the literature by findings on the biological effects mediated by Apigenin in cells lacking ERs [30]. To support this hypothesis, we here show that Apigenin modulates p38MAPK and ERK1/2 phosphorylation, but treatment with their inhibitors did not affect the expression of eNOS and MMP-9. However, we cannot exclude that Apigenin affects the expression and the activity of other molecules involved in the TNFα-induced endothelial dysfunction through p38MAPK and ERK1/2 modulation here described.
In conclusion, this study shows that Apigenin plays a protective role against endothelial dysfunction induced by TNFα. Our data clarified that Apigenin modulates multiple signalling pathways. Among these, the inhibition of Akt signalling through ER engagement plays a central role in reversing the TNFα-induced expression of eNOS and MMP-9. Both ‘classic’ ERs and GPR30 mediate the vaso-protective action of Apigenin, although to different extents. ERs have been implicated in protection from cardiovascular disease in women, and accordingly lack of oestrogen signalling is thought to be in part responsible for accelerated development of atherosclerosis in men and postmenopausal women [14, 15]. Therefore, having regard to its ability to bind to ERs, Apigenin may be considered an oestrogen-like molecule to potentially be used against the onset and progression of vascular diseases associated with endothelial dysfunction.
References
Vanhoutte PM, Shimokawa H, Tang EHC, Feletou M (2009) Endothelial dysfunction and vascular disease. Acta Physiol 196:193–222
Zhang H, Park Y, Wu J, Chen XP, Lee S, Yang J, Dellsperger KC, Zhang C (2009) Role of TNF-α in vascular dysfunction. Clin Sci 116:219–230
Vita JA (2005) Polyphenols and cardiovascular disease: effects on endothelial and platelet function. Am J Clin Nutr 81:292–297
Palmieri D, Pane B, Barisione C, Spinella G, Garibaldi S, Ghigliotti G, Brunelli C, Fulcheri E, Palombo D (2011) Resveratrol counteracts systemic and local inflammation involved in early abdominal aortic aneurysm development. J Surg Res 171:e237–e246
Aliakbarian B, Palmieri D, Casazza AA, Palombo D, Perego P (2012) Antioxidant activity and biological evaluation of olive pomace extract. Nat Prod Res. doi:10.1080/14786419.2012.660692
Shukla S, Gupta S (2010) Apigenin: a promising molecule for cancer prevention. Pharm Res 27:962–978
Benavente-Garca O, Castillo J (2008) Update on uses and properties of citrus flavonoids: new findings in anticancer, cardiovascular, and anti-inflammatory activity. J Agric Food Chem 56:6185–6205
Yamagata K, Miyashita A, Matsufuji H, Chino M (2010) Dietary flavonoid apigenin inhibits high glucose and tumor necrosis factor alpha-induced adhesion molecule expression in human endothelial cells. J Nutr Biochem 21:116–124
Yamagata K, Miyashita A, Chino M, Matsufuji H (2011) Apigenin inhibits tumor necrosis factor alpha plus high glucose-induced LOX-1 expression in human endothelial cells. Microvasc Res 81:60–67
Lee JH, Zhou HY, Cho SY, Kim YS, Lee YS, Jeong CS (2007) Anti-inflammatory mechanisms of apigenin: inhibition of cyclooxygenase-2 expression, adhesion of monocytes to human umbilical vein endothelial cells, and expression of cellular adhesion molecules. Arch Pharm Res 30:1318–1322
Long X, Fan M, Bigsby RM, Nephew KP (2008) Apigenin inhibits antiestrogen-resistant breast cancer cell growth through estrogen receptor-α-dependent and estrogen receptor-α-independent mechanisms. Mol Cancer Ther 7:2096–2108
Mak P, Leung YK, Tang WY, Harwood C, Ho SM (2006) Apigenin suppresses cancer cell growth through ERβ. Neoplasia 8:896–904
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signalling. Science 307:1625–1630
Mendelsohn ME, Karas RH (1999) The protective effects of estrogen on the cardiovascular system. N Engl J Med 340:1801–1811
Meyer MR, Schurr U, Barton M (2009) Erα, Erβ, and gpER: novel aspects of oestrogen receptor signalling in atherosclerosis. Cardiovasc Res 83:605–610
Palombo D, Maione M, Cifiello BI, Udini M, Maggio D, Lupo M (1999) Matrix metalloproteinases. Their role in degenerative chronic diseases of abdominal aorta. J Cardiovasc Surg 40:257–260
Thompson RW, Parks WC (2006) Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann N Y Acad Sci 800:157–174
Galis ZS, Sukhova GK, Lark MW, Libby P (1994) Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 94:2493–2503
Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C (2007) TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 27:1269–1275
Yang B, Rizzo V (2007) TNFa potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane raft and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 292:H954–H962
Paik DC, Ramey WG, Dillon J, Tilson MD (1997) The nitrite/elastin reaction: implication for in vivo degenerative effects. Connect Tissue Res 36:241–251
Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS (1996) Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro: implication for atherosclerotic plaque stability. J Clin Invest 98:2572–2579
Huang CH, Kuo PL, Hsu YL, Chang TT, Tseng HI, Chu YT, Kuo CH, Chen HN, Hung CH (2010) The natural flavonoid apigenin suppresses Th1- and Th2-related chemokine production by human monocyte THP-1 cells through mitogen-activated protein kinase pathways. J Med Food 13:391–398
Paoletti T, Fallarini S, Gugliesi F, Minassi A, Appendino G, Lombardi G (2009) Anti-inflammatory and vascular protective properties of 8-prenylapigenin. Eur J Pharmacol 620:120–130
Lin M, Lu SS, Wang AX, Qi XY, Zhao D, Wang ZH, Man MQ, Tu CX (2011) Apigenin attenuates dopamine-induced apoptosis in melanocytes via oxidative stress-related p38, c-Jun NH2-terminal kinase and Akt signaling. J Dermatol Sci 63:10–16
Lee WJ, Chen WK, Wang CJ, Lin WL, Tseng TH (2008) Apigenin inhibits HGF-promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and beta 4 integrin function in MDA-MB-231 breast cancer cells. Toxicol Appl Pharmacol 226:178–191
Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH (2005) Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J 19:342–353
Bersacchi R, Perrotta C, Bulotta S, Moncada S, Borghese N, Clementi E (2003) Activation of endothelial nitric-oxide synthase by tumor necrosis factor-α: a novel pathway involving sequential activation of neutral Sphingomyelinase, phosphatidylinositol-3′ kinase and Akt. Mol Pharmacol 63:886–895
Xing F, Jiang Y, Liu J, Zhao, Liu Z, Zeng Y (2006) Downregulation of human endothelial nitric oxide synthase promoter activity by p38 mitogen-activated protein kinase activation. Biochem Cell Bio 84:780–789
Lindenmeyer F, Li H, Menashi S, Soria C, Lu H (2001) Apigenin acts on the tumor cell invasion process and regulates protease production. Nutr Cancer 39:139–147
Acknowledgments
This study was supported by a grant of Compagnia di San Paolo Foundation to Prof. Domenico Palombo. We are grateful to Valentina Guani for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Palmieri, D., Perego, P. & Palombo, D. Apigenin inhibits the TNFα-induced expression of eNOS and MMP-9 via modulating Akt signalling through oestrogen receptor engagement. Mol Cell Biochem 371, 129–136 (2012). https://doi.org/10.1007/s11010-012-1429-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1429-1