
Abstract
Degradation and resynthesis of the extracellular matrix (ECM) are essential during tissue remodeling. Expansion of the vascular intima in atherosclerosis and restenosis following injury is dependent upon smooth muscle cell (SMC) proliferation and migration. The migration of SMC from media to intima critically depends on degradation of ECM protein by matrix metalloproteinases (MMPs). MMP inhibitors and eNOS gene transfer have been shown to inhibit SMC migration in vitro and neointima formation in vivo. Nitric oxide (NO) and cyclic-GMP have been implicated in the inhibition of VSMC migration. But, there are few studies addressing the role of NO signaling pathways on the expression of MMPs. Here we reported the involvement of cyclic-GMP-dependent protein kinase (PKG) (an important mediator of NO and cGMP signaling pathway in VSMC) on MMP-2 expression in rat aortic SMC. The goal of the present study was to gain insight into the possible involvement of PKG on MMP-2 in rat aortic SMC. MMP-2 protein and mRNA level and activity were downregulated in PKG-expressing cells as compared to PKG-deficient cells. In addition, the secretion of tissue inhibitor of metalloproteinase-2 (TIMP-2) was increased in PKG-expressing cells as compared to PKG-deficient cells. PKG-specific membrane permeable peptide inhibitor (DT-2) reverses the process. Interestingly, little or no changes of MMP-9 were observed throughout the study. Taken together our data suggest the possible role of PKG in the suppression of MMP-2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vascular remodeling in response to injury and inflammation is one of the hallmarks of vascular diseases such as arteriosclerosis and restenosos [1, 2]. The tissue response to injury includes the migration and proliferation of vascular smooth muscle cells (VSMC), the increased synthesis of extracellular matrix proteins, and the expression of matrix degrading proteases, the matrix metalloproteinases (MMPs) by VSMC [3, 4]. These biochemical changes in the phenotype of VSMC are part of the phenotypic modulation process that occurs in response to injury in vivo and to repetitive passaging of cultured VSMC in vitro [5].
There have been several reports that demonstrate that the inhibition of MMP production, or alternatively, inhibition of MMP activity, decreases VSMC migration, proliferation and neointimal growth in animal models of vascular injury [6, 7]. Nitric oxide (NO) released from endothelial cells or from NO donor drugs, inhibits VSMC proliferation, migration, and matrix protein synthesis [8–10]. Our laboratory has demonstrated that passaging adult rat aortic VSMC in culture or the application of a balloon catheter injury in vivo suppresses the expression of the downstream target of NO signaling, cGMP-dependent protein kinase (PKG), coincident with increased proliferation and expression of matrix proteins. Restoration of expression of PKG using either transfection methods or adenoviral gene transfer in vitro or in vivo inhibits matrix protein production and neointimal growth, respectively [11, 12]. These findings have led to the concept that NO signaling and activation of PKG suppress vascular lesion formation and inhibit vascular remodeling in response to injury and inflammation.
The role of the MMPs in vascular lesion formation and vascular diseases has been studied extensively. Two major forms of the MMP family, MMP-2 and MMP-9, are found in atherosclerotic lesions, suggesting a role for these two proteases in vascular wall remodeling [13, 14]. In addition, tissue inhibitors of metalloproteinase (TIMPs) are produced by VSMC [15], and other cell types to inhibit MMP activity thus attenuating matrix breakdown and remodeling. It has been shown that VSMC expressing at least one form of TIMP, the TIMP-1, inhibits neointimal formation in animal models of vascular injury [16]. There are few other reports on the role of NO signaling and TIMP activity in VSMC [17]. Given the important roles for MMPs and TIMPs in the turnover of vascular matrix and the remodeling of the vascular wall in response to injury, it is surprising that there are few studies addressing the role of NO signaling pathways on the expression of these substances. In this report, we have used primary, cultured rat aortic VSMC (both deficient in PKG expression and over expressing PKG) to determine the role of this NO signaling pathway on the expression of MMPs and TIMPs.
Materials and methods
Reagents
Fetal bovine serum (FBS), Dulbecco’s modified minimal essential medium (DMEM) were purchased from Gibco (Grand Island, NY). 8-para-cholorophenylthio-cGMP (8-CPT-cGMP) from Biolog (Bremen, Germany). 8-Bromoguanosine 3′,5′-cyclic monophosphate(8-Bromo-cGMP) from Sigma (Saint Louis, MO, USA) Polyclonal antibody to PKG-I was purchased from StressGen Biotechnology, Inc. (Victoria, BC, Canada). pVASP(Ser239) antibody was purchased from Cell signaling Technology Inc., USA. PKG specific peptide inhibitor (DT-2) is a kind gift from Dr. W R. Dostmann, University of Vermont, USA. Antibodies to MMP-2 and TIMP-2 and purified MMP-2 were purchased from Santa Cruz Biotec. Inc. (CA, USA).
Smooth muscle cell isolation and culture
In this study male Sprague–Dawley rats were used in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996) and in compliance with applicable laws and regulations. Animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of South Alabama. Rat aortic VSMC were isolated form the thoracic and abdominal aorta of the Sprague–Dawley male rat (150–200 g) as described previously [18]. In brief, the rats were sacrificed by CO2 inhalation, the aortas excised, and placed in “isolation medium” of DMEM containing 20 mM N-2-hydroxyethylpiperazine-N′2-ethanesulfonic acid, 1 mg/ml bovine serum albumin (BSA), 5 μg/ml amphotericin B, and 50 μg/ml gentamicin. After cleaning the tissues and removing adhering fat and connective tissue, aortas were placed in a digestion medium consisting of isolation medium plus 1 mg/ml elastase and 130 U/ml of collagenase for 10 min. The aortas were removed from the solution and rinsed to wash away endothelial and other non-adhering cells, and the tunicae adventitiae were removed as everted tubes using fine forceps. The remaining medial layer was minced and further digested for 1–2 h in a digestion medium containing isolation medium plus 200 U/ml collagenase until a single cell suspension was obtained. Cells were washed twice by low-speed centrifugation in isolation medium and placed in tissue culture medium containing DMEM, 10 % FBS, and 50 μg/ml gentamicin. Before any passage after isolation cells were grown up to confluence and used as primary cells for experiments. Cells were routinely plated in plastic tissue culture plates without any matrix coating in a 90 % air, 10 % CO2 humidified incubator for the first passage, and then transferred to a standard 95 % air, 5 % CO2 atmosphere for additional passages. For subculturing, cells were removed from the tissue culture plates using 0.05 % tris-buffered trypsin and split at a ratio of 1:5. Cells cultured under these conditions generally express low levels of PKG after passage 4–6 (less than 10 % of unpassaged cells). Primary smooth muscle cells were treated with 250 μM 8-Bromo-cGMP followed by serum deprived. Condition medium was then collected for gelatinolytic activity of MMP-2.
Cell transfection
The cDNA encoding bovine PKG-Iα was cloned into pcDNA-1 neo into the BamHI site, amplified and purified using a Wizard Maxi Prep. Five micro gram of vector containing PKG-1α cDNA was used to transfect rat aortic VSMC at passage 2 using 10 μl of transfectam reagent with precipitation of the DNA–liposome complex for 15 min at room temperature. The precipitate was added to the cell monolayer and the cells were incubated for 6 h at 37 °C in an atmosphere of 90 % air, 10 % CO2. The transfection was terminated by adding DMEM with 20 % FBS. Control treatments were transfection of a catalytically-inactive PKG-Iα (critical lysine mutant) or simply empty vector controls. Stably transfected cells were selected using 500 μg/ml G418. After isolation of colonies from 96-well plates, the transfected cell lines were maintained in 250 μg/ml G418 for ~8 passages following plating. For all subsequent experiments, cells were plated and allowed to attach overnight. Cells were then serum deprived for 24 h in DMEM containing 1 mg/ml BSA before experiments were performed. DT-2 was added to the culture medium and incubated before collecting condition medium and cells for western blot and gelatinolytic activity.
RNA isolation and RT-PCR
Total RNA was isolated from control and PKG stable transfected rat aortic VSMC using the Quiagen RNAEasy RNA isolation kit following the manufacturer’s instructions. The one step RT-PCR was performed using primer sets from the rat MMP-2 sequences [19]. The PCR product was analyzed in 1 % agarose gels and visualized using UV irradiation.
Affinity purification of MMPs
MMP-2 and MMP-9 were affinity purified according to the procedure described by Rajagopalon et al. [20]. In brief, 1 ml of conditioned medium from control and PKG-transfected cells was added to 100 μl of gelatin–agarose beads and the mixture was incubated for 1 h at 4 °C. The mixture was then centrifuged briefly and the beads were washed two times with buffer. The MMP was eluted from the washed beads using cold 10 % DMSO. The eluted MMP was applied to a polysulfone ultrafuge filter (30,000 MW exclusion) and centrifuged at 3500 rpm to remove DMSO. The filtered supernatant was used as the source of MMP.
Zymography
MMP-2 gelatinolytic activity was identified using an in-gel 10 % SDS-PAGE electrophoresis assay according to the procedure of [20]. Samples were applied to gels containing 1 mg/ml gelatin and electrophoresed. Following renaturation by exchanging SDS for Triton-X-100 (2.5 % Triton in 50 mM Tris–Cl, pH 7.5) two times for 25 min each, the gels were incubated overnight at 37 °C in 50 mM Tris–Cl, pH 7.5 containing 10 mM CaCl2 and 0.05 % Brij 35 detergent. Gels were stained with coomassie blue, partially destained, and photographed. The proteins having gelatinolytic activity were visualized as the area of lytic activity, i.e., a white band on a light blue background.
Western blot
Conditioned medium from transfected rat aortic VSMC was subjected to affinity purification for MMP as described. The fractions were applied to 10 % SDS–polyacrylamide gels and proteins separated by electrophoresis. The proteins were transferred to nitrocellulose, blocked in 5 % non-fat dried milk in TBS buffer for 1 h at room temperature, and incubated with primary antibody toward MMP-2 overnight. The next day, the blots were washed three times in TBS buffer, incubated in secondary antibody solutions, and bands were developed using chemilumenescence.
Northern blot
Total cellular RNA was isolated from the cultured cells using the RNA STAT-60 isolation reagent according to the manufacturer’s instructions (Tel-Test, Inc.). RNA (15 μg) was resolved on 1 % formaldehyde agarose gel, transferred to Nytran membrane, and UV crosslinked. After prehybridization in Quickhyb (Stratagene), the membrane was incubated with 32[P]-labeled PCR generated MMP-2 and TIMP-2 probes. Hybridization was carried out for 2–3 h at 65 °C. Membranes were then washed twice with 2× SSC in 0.2 % SDS, twice at room temperature, and again at 50 °C as needed. After washing the blots were exposed to kodak film and developed.
Morphology and microscopic analysis
To examine cells for MMP-2 expression using immunocytofluorescence microscopy, cells (50,000 cells/1 cm2 coverslip) were grown in DMEM containing 10 % FBS. For MMP-2 immunofluorescence, the medium was removed and replaced with DMEM in the absence of serum and incubated for 2 h at 37 °C. The cells were washed two times in PBS and fixed in 100 % methanol for 5 min at −20 °C. After fixation, cells were washed twice with PBS and incubated with 1 % BSA for 1 h at room temperature, then washed three times with PBS. Cells were then incubated for 1 h at room temperature with monoclonal anti-MMP-2 and anti-α-actin antibody diluted in 1 % BSA, washed three times with PBS, then incubated with FITC-conjugated anti-mouse IgG (1:50) for 1 h at room temperature. The cells were then washed three times in PBS and mounted in a slide containing one drop of mounting medium. The slides were sealed with white nail polish and examined using a Leica confocal fluorescence microscope equipped with Nikon camera. Nuclei were stained with 1 μg/ml DAPI in PBS 5 min at room temperature in same slides. Preimmune serum controls were performed using non-immune host serum. All experiments were repeated 3–4 times with similar results.
Migration assay
For SMC migration study, in vitro scratch or wound healing assay was performed [30]. In brief, SMC were cultured in monolayer and transfected with PKG cDNA and control cDNA. After transfection, wounds were made by scratching the monolayer with a 200 μl pipet tip, followed by washing with medium to remove the cell debris. Scratched regions were photographed immediately after injury (time 0) and after 24 h under an inverted microscope using Nikon Cool Pix 4500 digital camera attached with microscope. Images were taken from same plates each time point. The experiments were repeated three times with duplicate for each condition.
Statistical analysis
All experiments were repeated at least three times. Data represents are mean ± SD. Value of p < 0.05 were considered statistically significant.
Results
Characterization of PKG-expressing cells
The expression of PKG in adult rat aortic VSMC decreases with cell passage as described [21, 22]. In part, this appears to be due to disruptions in cell–cell contact that occurred with subculturing [3, 5] so that by passage 4–6, PKG-I expression at the protein level decreased. The effects of PKG expression on cell phenotype and morphology are shown in Fig. 1a, VSMC when transfected with PKG-1α took on a more contractile phenotype with cells demonstrating an elongated morphology and growing in parallel fashion similar to primary cells. PKG-deficient cells, on the other hand grew as rounded cells in a random fashion. Western blot (Fig. 1b) shows the differences in PKG expression in primary, passaged and PKG-transfected SMC. In order to evaluate the PKG activity in primary, passaged and PKG-transfected cells, we assessed PKG activity by using VASP phosphorylation using the anti-ser-239 VASP antibody. As shown (Fig. 1c) in primary and PKG-transfected cells activity is almost equal although less pVASP is seen in passaged cells compared with primary and PKG-transfected cells. Figure 1d showing more MMP-2 production by passaged control-transfected cells, however, almost undetectable in primary and PKG tranfected cells. As reported previously [11, 18], PKG expression increases the levels of contractile proteins (SMMHC, caldesomon) and decreases the levels of extracellular matrix proteins osteopontin, thrombospondin, and collagen 1.

Effect of PKG-1 over expression on cell phenotype. a Phenotypic difference among primary, PKG-deficient and PKG-transfected rat aortic SMC. b Western blot for PKG-1 levels in primary, non-transfected and PKG-transfected rat aortic SMC extracts. c Western blot for PKG activity by VASP phosphorylation in primary, non-transfected and PKG-transfected cells. d Gelatinolytic activity for MMP-2 in control-transfected, PKG-transfected, and early passaged VSMC
Effect of PKG expression on MMP-2 expression
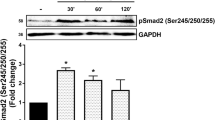
Cells expressing PKG-1 had a reduced level of MMP-2 mRNA (Fig. 2a, b) and protein (Fig. 2c) when compared to control-transfected cells. The RNA results were obtained using both RT-PCR and northern blot approaches. These results suggested that MMP-2 secretion was greater in the non-PKG-expressing cells compared to PKG-expressing cells as a result of a greater synthesis of MMP-2. In order to confirm that PKG inhibited MMP-2 activity in VSMC, zymogram analysis was performed using gels containing 1 mg/ml gelatin as the substrate. Expression of PKG-1 in rat aortic VSMC resulted in decrease in MMP-2 activity (Fig. 2d) in conditioned medium from the cultured cells. To demonstrate the importance of PKG we took another approach. We reported [31] prolonged treatment of cells with the cGMP analog, 8-Bromo-cGMP, down-regulated PKG via the ubiquitin priteasomal pathway. Figure 2 e showing that treatment of SMC with 250 μM 8-Bromo-cGMP increases the MMP-2 production. MMP-9 levels on the other hand were very low and did not appear to be different.

RT-PCR, northern blot and western blot analysis for MMP-2 in control and PKG-transfected aortic SMC. a Total RNA was isolated from the two cell phenotypes and RT-PCR was performed for MMP-2 by using specific primers. b Northern blot analysis for MMP-2 using a 32 [P]-labeled cDNA probe. c Western blot analysis for MMP-2 from whole cell lysate or conditioned media. Graph represents the MMP-2 band density. The values mean ± SD. d Gelatin zymography. Positions of MMP-9, MMP-2, and pro MMP-2 indicated. e Gelatinolytic activity of MMP-2 in 8-Bromo-cGMP untreated and treated SMC. Standards were run along with sample in each experiment
TIMP expression
Tissue inhibitors of metalloproteinases, or TIMPs, decrease the activity of the secreted MMP enzymes by binding tightly to their catalytic sites. As shown (Fig. 3a, b) PKG-1 expression increased both RNA and protein (Fig. 3c) expression for the TIMP-2 isoform in rat aortic VSMC. Protein was detected in the media from PKG-transfected cells, but not from the control-transfected cells.

RT-PCR, northern blot and western blot analysis for TIMP-2. a RT-PCR for TIMP-2 in rat aortic SMC. b Northern blot for TIMP-2. c Western blot for TIMP-2 in condition media of rat aortic VSMC. Graph shows average TIMP-2 band density. The values mean ± SD
Immunocytofluorescence analysis of MMP-2 levels in VSMC
Because MMP-2 is synthesized as a precursor protein in VSMC, it was of interest to examine the effects of PKG-1 transfection on the expression levels and the distribution of MMP-2 in the cells. MMP-2 fluorescence (Fig. 4b) was widely distributed in non-PKG-transfected cells compared to a very sparse distribution of stain in PKG-expressing cells. Smooth muscle-specific actin staining was also performed to assess cellular distribution of MMP-2 in control and PKG-expressing cells. As shown in Fig. 4c, PKG-expressing cells demonstrated a higher organization of actin into filaments compared with control-transfected cells, although the intensity of staining did not appear to be dramatically different between the two cell types. Thus, PKG affected both actin filament formation and MMP-2 expression and distribution in rat aortic VSMC.

Immunocytofluorescence for MMP-2 expression and α-actin in control and PKG-transfected cells. Preimmune serum (a), anti-MMP-2 (b), and anti alpha actin (c)
Effect of DT-2 on MMP-2 expression
To further evaluate the possible involvement of PKG on MMP-2 inhibition, we used DT-2 [24]. Following treatment with 10 μM DT-2, both MMP-2 secretion and expression were increased as compare to control, shown by gelatinolytic activity (Fig. 5a) and western blot analysis (Fig. 5b). This data may suggest a role of PKG on MMP-2 inhibition.

Effect of DT-2 on MMP-2 secretion and expression in PKG-expressing SMCs. Cells were treated with or with out 10 μM DT-2 (a PKG-specific inhibitor) and conditioned media was collected for MMP-2 gelatinolytic activity (a) and cells from same plate was used to do western blot (b) for MMP-2. Graphs represent average band density for gelatinolytic activity and western blot
Effect of MMP-2 inhibition on SMC migration
We next tested PKG expression on SMC migration by the wound healing assay. Figure 6 shows that in PKG-expressing cells, migration was significantly lower as compare to control. As shown in Fig. 5a, b, the level of MMP-2 expression and production was also significantly lower in PKG cells as compare to control. This observation may indicate an important correlation between PKG expression and MMP-2 inhibition, resulting in the inhibition of SMC migration, which is the main event during neointima formation after an injury or inflamation in the arterial wall.

Migration of SMC in control and PKG-transfected cells. Wound healing assay was performed as describe in methods. Representative images were taken at 0 and 24 h after making the scratch in the cell monolayer. Graph represents the distance (cm) between the line in control cells and PKG cells after 0 and 24 h. Less distance indicated more migration
Discussion
In the present study, we have characterized expression and activity of MMPs in cultured rat aortic VSMC in response transfection of PKG-Iα. Passaged cells transfected with empty vector express only ~20 % of the normal level of PKG-1 as determined by western blotting, and express lower levels of several contractile proteins [23]. These results suggest that PKG is capable of regulating some aspects of smooth muscle-specific gene expression, although the mechanisms are obscure. In addition to effects on contractile protein expression, we and others [11, 29] have found that PKG-1 transfection in VSMC or mesangial cells inhibits the expression of extracellular matrix proteins such as collagen, thrombospondin-1, and osteopontin. These findings are also consistent with a role for NO/cGMP signaling in preventing phenotypic modulation of cultured rat aortic VSMC to a more synthetic and fibroproliferative phenotype [24].
It has been reported by several laboratories [25] that VSMC or those present in atherosclerotic lesions express and secrete abundant levels of MMPs. MMP expression occurs when matrix turnover is necessary for cell migration, proliferation, angiogenesis, and wound healing, and several MMPs are found in vascular lesions [26]. In vivo studies suggest that remodeling that occurs in arterial lesions in response to injury is due in part to the increased production of MMPs necessary for tissue breakdown and resynthesis. In particular, MMP-2 and MMP-9 have been shown to be present in human arterial lesions and in animal models of restenosis [27, 28].
In the current study, we found that highly passaged cultured rat aortic VSMC synthesize and secrete both MMP-2 and to a lesser extent MMP-9. Restoration of PKG expression to the cells had little effect on the expression of MMP-9, but dramatically inhibited the expression and secretion of MMP-2. Previous studies [28] have demonstrated that both MMPs are increased during VSMC growth in rabbit aortic explants in vitro [14] and in balloon injured rat carotid artery in vivo. Our study also demonstrates that the MMP-2 expression which is produced by VSMC in a more synthetic proliferative phenotype is inhibited by PKG.
In addition, PKG-1 expression stimulates the level of TIMP-2 in rat aortic VSMC. This is the first report of an effect of PKG on this protein, and brings a new insight to the role of MMPs in response in arterial injury, and possible mechanisms whereby PKG inhibits matrix synthesis during vascular remodeling. TIMP proteins which suppress MMP activity, reduces matrix production and turnover, thus the expression of major TIMP isoform TIMP-2, in addition to the effect of PKG-1 on MMP-2 levels result in a net decrease in matrix turnover.
Interestingly, a previous report [16] demonstrated that MMP inhibitors only partially inhibit neointima formation, whereas over expression of TIMP-1 by adenoviral mediated gene transfer inhibited smooth muscle cell migration and reduced neontimal hyperplasia in the rat model of vascular balloon injury. Our results suggest that the effect of PKG on TIMP-2 expression and MMP-2 supression in VSMC are important mechanisms by which PKG inhibits SMC proliferation and migration and thus inhibition of neointima formation. 8-Bromo-cGMP induced PKG down regulation [31] in VSMC also causes increased production of MMP-2, which may indicate a role of PKG.
PKG inhibition by DT-2 thus increases MMP-2 expression, which introduces added evidence of a role of PKG activity on MMP-2. PKG silencing in freshly isolated SMC via siRNA mediated knockdown and in vivo study might provide very important information. The actual molecular mechanism by which PKG-1 affects the expression of these proteins is not known and is subjected to future studies.
References
Schwartz SM, Campbell GR, Campbell JH (1986) Replication of SMC in vascular diseases. Circ Res 58:427–444
Ross R (1993) The pathogenesis of atherosclerosis, a perspective for the 1990s. Nature 362:801–809
Southgate KM, Davies M, Booth RFG, Newby AC (1992) Involvement of extracellular-matrix degrading metalloproteinase in rabbit aortic smooth muscle cell proliferation. Biochem J 288:93–99
Pauly RR, Passaniti A, Bilato C, Monticone R, Cheng L, Papadopoulos N, Gluzband YA, Smith I, Weinstein C, Lakatta EG, Crow M (1994) Migration of vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ Res 75:41–54
Cornwell TL, Soff GA, Traynor AE, Lincoln TM (1994) Regulation of the expression of cGMP dependent protein kinase by cell density in vascular smooth muscle cells. J Vasc Res 31:330–337
Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW (1996) Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol 16:28–33
Bendeck MP, Irvin I, Reidy MA (1996) Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not noontime thickening after arterial injury. Circ Res 8(1):33–43
Garg UC, Hassid A (1989) Nitric oxide generating vasodilators and 8-Bromo-cyclic guanosine monophosphate inhibits mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83:1774–1777
Nakaki T, Nakayama M, Kato R (1990) Inhibition of nitric oxide and nitric oxide producing vasodilators of DNA synthesis in vascular smooth muscle cells. Eur J Pharmacol 189:347–353
Cayatte AJ, Palacino JJ, Horten K, Cohen RA (1994) Chronic inhibition of nitric oxide production accelerates neointima formation and impairs endothelial function in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol 14:753–759
Dey NB, Boerth NJ, Murphy- Ullrich, Chang PL, Prince CW, Lincoln TM (1998) Cyclic GMP dependent protein kinase inhibits osteopontin and thrombospondin production in rat aortic smooth muscle cells. Circ Res 82:139–146
Anderson PG, Boerth NJ, Liu M, McNamara DB, Cornwell TL, Lincoln TM (2000) Cyclic GMP-dependent protein kinase expression in coronary arterial smooth muscle in response to balloon catheter injury. Arterioscler Thromb Vasc Biol 20:2192–2197
Benedick MP, Irvine C, Reidy MA (1996) Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointima thickening after arterial injury. Circ Res 78:38–43
Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW (1996) Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol 16:28–33
Forough R, Koyama N, Hassenstab D, Lea H, Clowes M, Nikkari ST, Clowes AW (1996) Over expression of tissue inhibitor of matrix metalloproteinase-1 inhibits vascular smooth muscle cell function in vitro and in vivo. Circ Res 79(4):812–820
George SJ, Johnson JL, Angelini GD, Newby AC, Baker AH (1998) Adenovirus mediated gene transfer of the human TIMP-1 gene inhibits smooth muscle cell muscle migration and neontima formation in human saphenous vein. Hum Gene Ther 9(6):867–877
Gurjar MV, Sharma RV, Bhalla RC (1999) eNOS gene transfer inhibits smooth muscle cell migration and MMP-2 and MMP-9 activity. Arterioscler Thromb Vasc Biol 19:2871–2877
Boerth NJ, Dey NB, Cornwell TL, Lincoln TM (1997) Cyclic GMP dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res 34(4):245–259
Marti HP, McNeil L, Davies M, Martin J, Lovett DH (1993) Homology cloning of rat 72 KDa type IV collagenase: cytokine and second messenger inducibility in glomerular mesangial cells. Biochem J 291(pt.2):441–446
Rajagopalon S, Meng P, Ramasamy S, Harrison DG, Galis Z (1996) Reactive oxygen species produced by macrophage-derived foam cells regulates the activity of vascular matrix metalloproteinase in vitro; implications for atherosclerotic plaque stability. J Clin Invest 98(11):2572–2579
Renart J, Reiser J, Stark GR (1979) Transfer of protein from gel to diazobenzyloxymethyl paper and detection with antisera: a method for studying antibody specificity and antigen structure. Proc Natl Acad Sci USA 76(7):3116–3120
Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts JD Jr, Filippov G, Janssens SP, Rosenzweig A, Bloch KD (1998) Adenovirus mediated gene transfer of a cGMP-dependent protein kinase increases the sensitivity of vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem 273:34263–34271
Cornwell Tl, Lincoln TM (1989) Regulation of intracellular calcium levels in cultured vascular smooth muscle cells: reduction of ca+ by atriopeptin and 8-bromo cyclic GMP id mediated by cyclic GMP-dependent protein kinase. J Biol Chem 264:1146–1155
Dey NB, Foley KF, Lincoln TM, Dostmann WR (2005) Inhibition of cGMP-dependent protein kinase reverses phenotypic modulation of vascular smoth muscle cells. J Cardiovasc Pharmacol 45(4):404–413
Galis ZS, Sukhova GK, Lark MW, Libby P (1994) Increased expression of matrix metalloproteinase and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 94:2493–2503
Li Z, Li L, Zielke HR, Cheng L, Xiao R, Crow MT et al (1996) Increased Expression of 72 KD type IV collagenase (MMP-2) in human aortic atherosclerotic lesions. Am J Pathol 148:121–128
Lijnen HR, Soloway P, Collen D (1999) Tissue inhibitor of Matrix metalloproteinase-1 impairs arterial neointima formation after vascular injury in mice. Circ Res 85:1186–1191
Baron AV, Frisdal E, Eddahibi S, Deprez I, Baker AH, Newby AC et al (2000) Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer or doxycycline aggravates pulmonary hypertension in rats. Circ Res 87:418–425
Wang S, Wu X, Lincoln TM, Ullrich JEM (2003) Expression of constitutively active c-GMP dependent protein kinase prevents glucose stimulation of thrombospondin-1 expression and TGF-α activity. Diabetes 52:2144–2150
Chen KC, Wang YS, Hu CY, Chang LYC, Dai CY, Juo SHH (2011) OxLDL up- regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular disease. FASEB J 25:1718–1728
Dey NB, Busch JL, Francis SH, Corbin JD, Lincoln TM (2009) Cyclic GMP specifically suppresses Type-1α cGMP-dependent protein kinase expression by ubiquitination. Cell Signal 21(6):859–866
Acknowledgments
This work was supported by a grant from the NIH (HL66164).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dey, N.B., Lincoln, T.M. Possible involvement of Cyclic-GMP-dependent protein kinase on matrix metalloproteinase-2 expression in rat aortic smooth muscle cells. Mol Cell Biochem 368, 27–35 (2012). https://doi.org/10.1007/s11010-012-1339-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1339-2