
Abstract
Phytoestrogens are known to prevent tumor induction. But their molecular mechanisms of action are still unknown. This study aimed to examine the effect of apigenin on proliferation and apoptosis in HER2-expressing breast cancer cells. In our experiments, apigenin inhibited the proliferation of MCF-7 vec and MCF-7 HER2 cells. This growth inhibition was accompanied with an increase of sub G0/G1 apoptotic fractions. Overexpression of HER2 did not confer resistance to apigenin in MCF-7 cells. Apigenin-induced extrinsic apoptosis pathway up-regulating the levels of cleaved caspase-8, and inducing the cleavage of poly (ADP-ribose) polymerase, whereas apigenin did not induce apoptosis via intrinsic mitochondrial apoptosis pathway since this compound did not decrease mitochondrial membrane potential maintaining red fluorescence and did not affect the levels of B-cell lymphoma 2 (BCL2) and Bcl-2-associated X protein. Moreover, apigenin reduced the tyrosine phosphorylation of HER2 (phospho-HER2 level) in MCF-7 HER2 cells, and up-regulated the levels of p53, phospho-p53 and p21 in MCF-7 vec and MCF-7 HER2 cells. This suggests that apigenin induces apoptosis through p53-dependent pathway. Apigenin also reduced the expression of phospho-JAK1 and phospho-STAT3 and decreased STAT3-dependent luciferase reporter gene activity in MCF-7 vec and MCF-7 HER2 cells. Apigenin decreased the phosphorylation level of IκBα in the cytosol, and abrogated the nuclear translocation of p65 within the nucleus suggesting that it blocks the activation of NFκB signaling pathway in MCF-7 vec and MCF-7 HER2 cells. Our study indicates that apigenin could be a potential useful compound to prevent or treat HER2-overexpressing breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Apoptosis, or programmed cell death (PCD), plays an important role in various biological systems, such as embryonic development, cell turnover, and immune response against tumorigenic or virus-infected cells [1]. When apoptosis occurs, different cell morphological changes including the shrinkage of cell and nuclei, membrane blebbing, loss of cell membrane asymmetry and attachment, nuclear fragmentation, and chromatin condensation appear [2]. Apoptosis differs from necrosis in its morphological and biochemical features. In most tumor cells, apoptosis occurs via two different signaling pathways: the extrinsic and the intrinsic apoptosis pathways. The extrinsic pathway is related to the activation of the death receptors, such as Fas and tumor necrosis factor receptors (TNFR). Death domains (DD) of Fas are oligomerized and recruit Fas-associated death domain (FADD) and procaspase-8 to form death-inducing signaling complex (DISC). Procaspase-8 is cleaved and activated and released from the DISC into the cytoplasm where it activates caspase-3 to induce apoptosis [1, 3, 4]. The intrinsic pathway is related to changes in mitochondrial membrane potential (ΔΨm) and mitochondrial permeability transition, resulting in mitochondrial release of apoptogenic factors such as cytochrome c and apoptosis-inducing factor (AIF) into the cytoplasm [1, 5]. Cytochrome c binds to APAF1 and recruits procaspase-9 to form an apoptosome; caspase-9 activates effector caspases such as caspase-3 to induce apoptosis [4]. Caspase-3 from both extrinsic and intrinsic pathways is responsible for the cleavage of poly (ADP-ribose) polymerase (PARP) during cell death [6]. Extracellular signal-regulated kinases (ERK) pathway and Nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling pathway can inhibit this apoptotic signaling [4]. The cross talk between the extrinsic apoptosis pathway and the intrinsic apoptosis pathway exists; caspase-8 can cleave BID, a death-inducing member of the BCL2 family. The truncated BH3-interacting domain death agonist (BID) translocates to the mitochondria and induces cytochrome c release which subsequently results in caspase-9-dependent activation [1, 7]. Recently, it was reported that mitochondrial carrier homolog 2 (MTCH2) is a novel protein which acts as a receptor-like protein for the truncated BID in the outer membrane of mitochondria [8]. In addition, Smac, a mitochondrial protein that interacts with inhibitor of apoptosis proteins (IAPs) is released into the cytoplasm to inhibit the caspase-binding activity of IAPs upon apoptosis stimuli [9]. Proteasome inhibitors are known to induce apoptosis in melanoma cells [10]. They induce apoptosis suppressing the transcription factor FoxM1 and up-regulating pro-apoptotic protein, Noxa [10].
Phytoestrogens are phenolic compounds derived from plants and that structurally mimic the principal mammalian estrogen (17β-estradiol), and bind to the estrogen receptor (ER) [11–13]. There is also evidence that phytoestrogens have hormone-independent activities [13]. Phytoestrogens are suggested to induce the prevention or the growth suppression of various cancers including breast [14, 15], prostate [16, 17], colorectal [18], and bladder cancer [19]. Phytoestrogens (genistein and daidzein) have been reported to have the ability to reverse DNA hypermethylation in cancer cell lines [20]. Genistein is known to inhibit proliferation of colon cancer cells by attenuating a negative effect of EGF on tumor suppressor FOXO3 activity [21]. Apigenin, naturally occurring flavonoids is commonly presented in many fruits and vegetables, including parsley, onions, orange, tea, chamomile, wheat sprouts, and in some seasonings [22, 23]. Apigenin is known to have potent anti-inflammatory and anti-carcinogenic effect in various animal tumor model systems [23–25]. Apigenin also induces growth inhibition and apoptosis in a variety of cancer cell lines including breast [26], lung [27], colon [28, 29], prostate [22], leukemia [30], and pancreatic [31] cells.
The human epidermal growth factor receptor 2 (HER2) oncogene encodes a 185-kDa transmembrane receptor tyrosine kinase [32]. HER2 is overexpressed in breast cancer and other cancers [32] and HER2 overexpression is associated with poor disease-free survival [33]. HER2-positive breast carcinomas are likely to be more aggressive and fast growing than other types of breast cancer [34]. HER2-positive breast cancer patients are also less responsive to hormone treatment and have a shorter duration of response than HER2-negative breast cancer patients [35]. Currently, trastuzumab (Herceptin) is one of the key drugs in the treatment strategy for HER2-positive breast cancer [34]. It works against cancer by maximizing the expression and inhibitory effect of p27, which leads to cell cycle G1 arrest and growth inhibition [36]. However, many women do not respond to trastuzumab or develop resistance [37]. This has resulted in significant efforts to find other compounds which could effectively treat HER2-overexpressing breast cancer.
In this study, we investigated whether apigenin displays growth-suppressive activity on HER2-overexpressing breast cancer cells. For this purpose, we tested the effects of apigenin on proliferation and apoptosis of MCF-7 human breast cancer cells engineered to overexpress oncogenic HER2 (MCF-7 HER2) and control vector cells (MCF-7 vec). We also investigated the mechanism by which apigenin regulates the growth of MCF-7 vec and MCF-7 HER2 cells analyzing the cell cycle and measuring the levels of apoptotic molecules and intracellular signaling molecules. We also verified whether apigenin inhibits STAT3 or NFκB signaling pathway, leading to growth suppression of HER2-expressing breast cancer cells.
Nutrition of soybean, tofu, vegetables, fruits, leaves, and grains (rich in phytoestrogens) is well studied and appreciated. Phytoestrogen nutrition is known to reduce menopausal symptoms, risk of coronary heart disease, osteoporosis, help lower blood cholesterol and blood pressure levels, and protect against various types of cancer. Since we report here that apigenin may suppress HER2-positive breast cancer, this study advances human nutrition.
Materials and methods
Compounds
Apigenin (4′,5,7-trihydroxyflavone), 17β-estradiol (E2), 4-hydroxytamoxifen (4-OH-TAM), and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). These compounds were dissolved in dimethyl sulfoxide (DMSO) or ethanol, and the final concentration of DMSO or ethanol in the controls and each sample did not exceed 0.1 %. We found that 0.1 % DMSO or ethanol did not affect the cell growth rate as compared to 0 % DMSO or ethanol (no treatment) in breast cancer cells (data not shown). JC-1 was obtained from Molecular Probes (Invitrogen, Carlsbad, CA, USA).
Cell cultures
MCF-7 and SK-BR-3 human breast cancer cells (American Type Culture Collection, ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 50 U/ml penicillin, 50 mg/ml streptomycin and 10 % FBS (Welgene, Daegu, Korea) at 37 °C in an atmosphere of 5 % CO2. MCF-7 vec and MCF-7 HER2 cells were generated as described elsewhere [38]. In brief, 293T human kidney cells were transfected with pBMN-vec-GFP or pBMN-HER2-GFP, pCMV-VSVG (encoding vesicular stomatitis virus glycoprotein), and pCMV-gag-pol (containing the Moloney murine leukemia virus gag and pol genes). At 48 h post-transfection, retroviral particles were collected from 293T cell media and concentrated using amicon ultra (Millipore, Billerica, MA). MCF-7 cells were treated with the concentrated retrovirus particles and 8 μg/ml of polybrane (Sigma-Aldrich, St. Louis, MO, USA). After 48 h, the cells were selected by using flow cytometry.
Antibodies
Primary antibodies directed against FAS, cleaved caspase-8, PARP, HER2, phospho-HER2 (Tyr1248), AKT, phospho-AKT (Ser473), phospho-MDM2 (Ser166), and phospho-IκBα (Ser32/36) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). BCL2, BAX, p53, phospho-p53 (Ser15), p21, MDM2, ERK, phospho-ERK (Tyr204), JNK, phospho-JNK (Thr183/185), p38, phospho-p38 (Thr180/Tyr182), NFκB, and Lamin B were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). BCL-XL and Rb were purchased from BD Biosciences (Franklin Lakes, NJ, USA). STAT3, phospho-STAT3 (Tyr705), phospho-JAK1 (Tyr1022/Tyr1023), and IκBα were obtained from Upstate-Millipore (Billerica, MA, USA). Tubulin was from Sigma-Aldrich. Horseradish peroxidase (HRP)-conjugated secondary antibodies (mouse and rabbit) were purchased from Calbiochem (San Diego, CA, USA) and anti-goat secondary antibody was from Jackson ImmunoResearch (West Grove, PA, USA).
Cell proliferation assay
Cells were seeded in 12-well culture plates at a density of 5 × 104 cells/well. After the cells were exposed to different concentrations of apigenin (0–40 μM) and incubated for 3 days, the cells were harvested by trypsinization, resuspended in 1–2 ml of medium, and counted using a hemocytometer.
MTT assay
Cells were seeded in 96-multiwell culture plates at a density of 2 × 103 cells/well and incubated for 24 h at 37 °C. Then, cells were treated with different concentrations of apigenin (0–40 μM) for 24, 48, or 72 h. After incubation, MTT reagents (0.5 mg/ml) were added to the each well and the plates were incubated in the dark at 37 °C for 2 h. Medium was removed, formazan was dissolved in DMSO, and optical density was measured at 570 nm using an ELISA plate reader.
Cell cycle analyses by flow cytometry
Cells were harvested with 0.25 % trypsin and washed once with phosphate buffered saline (PBS). After centrifugation, the cells were fixed in 95 % cold ethanol with 0.5 % Tween-20, and stored at −20 °C for at least 30 min. The cells were incubated in 50 μg/ml of propidium iodide (PI) (including 1 % of sodium citrate and 50 μg/ml of RNase A) at room temperature in the dark for 30 min. The analysis of apoptotic cells were performed on a FACScan flow cytometer (Becton–Dickinson, Mountain View, CA, USA) and the data were analyzed using CellQuest software.
Analysis of mitochondrial transmembrane potential (ΔΨm)
Cells were seeded at a density of 1 × 106 cells/dish in 100 mm dishes and incubated for 24 h at 37 °C. After stabilization, the cells were treated with 40 μM apigenin and DMSO for 72 h. After harvest by treatment of trypsin–EDTA, the cells were washed with cold PBS, centrifuged at 1,500 rpm for 5 min and stained with 4 μg/ml JC-1 for 15 min at 37 °C in the dark. The data were analyzed by FACSCalibur flow cytometry (BD Biosciences) measuring the green fluorescence and red fluorescence at 514/529 nm (FL-1) and 585/590 nm (FL-2), respectively.
Preparation of cytosolic and nuclear protein
Cells were incubated in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol (DTT), and 0.5 % NP40). The cells were stood on ice for 5 min and then centrifuged at 3,000 rpm for 5 min. The supernatant was collected as cytosol extract. The pellet was then lysed with Buffer B (5 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and 26 % glycerol), and centrifuged at 5,000 rpm for 5 min at 4 °C then 4.6 M NaCl was added to give 300 mM NaCl. The pellet was homogenized with 20 full strokes in Dounce or glass homogenizer on ice. The pellet was incubated on ice for 30 min, and then centrifuged at 24,000 rpm for 20 min at 4 °C. Nuclear proteins were obtained from the supernatant.
Western blot analysis
Cells were lysed in modified RIPA buffer (150 mM NaCl, 1 % NP-40, 0.5 % deoxycholate, 0.1 % SDS, 50 mM Tris (pH 8.0), 1 mM EDTA, 1 mM phenylmethylsulfonylfluoride (PMSF), 1 mM NaF, 1 mM Na3VO4, and protease inhibitor mixture). The lysates were cleared by centrifugation at 13,000 rpm for 15 min and the supernatants were collected. The protein concentration was quantified using a Bio-Rad Bradford protein assay (Bio-rad, Hercules, CA, USA). Equal amounts of protein lysates were used for western blot analyses with the indicated antibodies. Immunoreactive protein bands were detected with an EZ-Western Detection kit (Daeillab service Co, Ltd, Seoul, Korea).
Immunocytochemistry
Cells (2 × 104 cells/well) were seeded in 8-well chamber slides, incubated for 24 h at 37 °C and treated with apigenin (40 μM) for another 24 h. The cells were fixed with 4 % paraformaldehyde for 30 min and treated with 3 % hydrogen peroxide (H2O2) in methanol for 20 min to quench endogenous peroxidase activity. The cells were washed with PBS, blocked with 5 % BSA in PBS for 1 h and incubated with anti-NF-κB (p65) primary antibody (1:200 dilution) overnight at 4 °C. After washing with PBS, the cells were incubated with anti-mouse biotin-conjugated secondary antibody for 1 h at room temperature. Then, the cells were treated with ABC reagent for 30 min at 4 °C and stained with diaminobenzidine tetrachloride (DAB) and hematoxylin. The cells were mounted with mounting medium and subsequently analyzed by microscopy.
STAT3 luciferase reporter assay
To measure STAT3 transcriptional activity, we used Hep3B-pSTAT3-luciferase stable cell line. This cell line was a kind gift of Dr. Lee SG (Cancer preventive material development research center, college of oriental medicine, Kyung Hee University). For the assay, cells were plated and allowed to attach by overnight incubation at 37 °C. After treatment with apigenin (40 μM), IL-6 (10 ng/ml) or IL-6 and apigenin for 24 h, cells were lysed using a passive lysis buffer (promega, Madison, WI, USA). Luciferase assays were performed using a dual-luciferase assay kit according to the manufacturer’s instructions (Promega). Finally, luciferase activities were determined using a luminometer (BMG Labtech, Ortenberg, Germany).
Statistical analysis
The experiments were performed in triplicate. The data for cell proliferation assay, MTT assay, and STAT3 luciferase reporter assay are expressed as mean ± standard deviation. Standard deviations for all measured biological parameters are displayed in the appropriate figures. Student’s t test was used for single variable comparisons, a p value <0.05 being considered as statistically significant.
Results
Apigenin suppresses the growth of MCF-7 vec and MCF-7 HER2 breast cancer cells
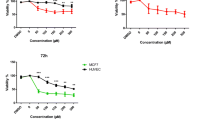
The effects of apigenin on cell growth were measured by cell proliferation assay and/or MTT assay in MCF-7 and SK-BR-3 cell lines. As reported, SK-BR-3 cells naturally overexpress oncogenic HER2 while MCF-7 HER2 cells are engineered to overexpress HER2. As seen in Fig. 1a, apigenin inhibited significantly cell proliferation in a dose-dependent manner (0–40 μM) in both MCF-7 vec and MCF-7 HER2 cells after 72 h of treatment (proliferation assay). Apigenin also suppressed the growth of SK-BR-3 cells in a dose-dependent manner (0–40 μM) after 72 h of treatment. It should be noted that SK-BR-3 cells were less sensitive to apigenin at the level of cell growth inhibition as compared to MCF-7 HER2 cells. That is why we chose MCF-7 HER2 cells to investigate the mechanism by which apigenin induces apoptosis in HER2-overexpressing breast cancer cells. We also chose MCF-7 HER2 cells for the experiments to study the implication of ERs in our system since SK-BR-3 cells do not contain ER. As seen in Fig. 1b, MTT assay demonstrated that apigenin suppressed cell growth rate in a time-dependent manner in both MCF-7 vec and MCF-7 HER2 cells. It seems that proliferation assay is more sensitive than MTT assay at measuring the intensity of cell growth inhibition as seen in Fig. 1. We expected that MCF-7 HER2 cells would exhibit resistance to apigenin due to the presence of the HER2 oncogene. However, we did not observe such resistance in our experiments. Overexpression of HER2 did not confer resistance to apigenin in MCF-7 cells. To verify implication of ERs in the pro-apoptotic action of apigenin, we treated the MCF-7 cells (ER α-positive) with ER α agonist (E2) and antagonist (4-OH-TAM) and performed proliferation assay (Fig. 1c). We found that E2 increased the proliferation of MCF-7 vec and MCF-7 HER2 cells while 4-OH-TAM decreased the growth of MCF-7 vec and MCF-7 HER2 cells. Apigenin abrogated E2-induced cell growth suggesting that it inhibits cell growth promotion induced by ER-dependent mechanism. It seems that apigenin induces apoptosis by both ER-dependent and -independent manner.

Effect of apigenin on the growth of MCF-7 vec and MCF-7 HER2 cells. a MCF-7 vec, MCF-7 HER2, and SK-BR-3 cells were treated with different doses of apigenin (0–40 μM). After 72 h, the cell viability was assessed with proliferation assay. b MCF-7 vec and MCF-7 HER2 cells were treated with different doses of apigenin (0–40 μM). Relative cell growth rate was measured by MTT assay after 24, 48, and 72 h. The value from vehicle-treated cells was set to 100 % and relative decrease in the cell viability by apigenin treatment was expressed as a percentage of the control. c MCF-7 vec and MCF-7 HER2 cells were treated with DMSO, E2 (0.01 μM) or 4-OH-TAM (0.1 μM) in the presence or absence of apigenin (40 μM). After 72 h, the cell viability was assessed with proliferation assay. Data are shown as the mean of three independent experiments [error bars are mean ± standard deviation (SD)] (*p < 0.05, **p < 0.01, ***p < 0.001)
Moreover, the growth inhibition induced by apigenin was verified by microscopic observation. The results in Fig. 2 show that apigenin effectively inhibited the growth rate of both MCF-7 vec and MCF-7 HER2 monolayer cells after 72 h treatment. Of note, apigenin also induced morphological changes in the cells after exposure (Fig. 2).

Apigenin inhibited the proliferation of MCF-7 vec and MCF-7 HER2 cells. MCF-7 vec and MCF-7 HER2 cells were seeded at a density of 1 × 106 cells on a 100 mm dish. The following day, the cells were treated with apigenin (0–40 μM) for 72 h and photographed by phase contrast microscopy (magnification of ×40). Control groups of cells were treated with DMSO alone
The growth-suppressive activity of apigenin is accompanied by an increase in sub G0/G1 apoptotic fractions in MCF-7 vec and MCF-7 HER2 breast cancer cells
To investigate whether apigenin inhibited cell proliferation through changes in cell cycle progression, the effect of apigenin on the cell cycle profile was assessed in MCF-7 vec and MCF-7 HER2 cells. For that purpose, cells were treated with 40 μM apigenin for 72 h and then analyzed for cell cycle location by flow cytometry. The data demonstrated that apigenin induced an increase in the sub G0/G1 apoptotic fractions in both MCF-7 vec and MCF-7 HER2 cells (Fig. 3a, b).


Effect of apigenin on the cell cycle and sub G0/G1 apoptotic fractions of MCF-7 vec and MCF-7 HER2 cells. a MCF-7 vec and b MCF-7 HER2 cells were treated with apigenin (0–40 μM) and the cells were fixed at 72 h for flow cytometry analyses. PI-labeled nuclei were analyzed for DNA content. Sub G0/G1 apoptotic fractions, G1, S and G2/M phase fractions were quantified from DNA histograms data
Apigenin does not induce apoptosis via the intrinsic mitochondrial apoptosis pathway in MCF-7 vec and MCF-7 HER2 breast cancer cells
Next, we investigated whether apoptosis induced by apigenin occurs via intrinsic mitochondrial apoptosis pathway in MCF-7 vec and MCF-7 HER2 cells. For that purpose, we measured the loss of mitochondrial transmembrane potential (ΔΨm) within the cells using JC-1. JC-1 is able to selectively enter mitochondria and reversibly transforms color from red to green when the membrane potential decreases. In non-apoptotic cells with high mitochondrial ΔΨm, JC-1 spontaneously forms complexes known as J-aggregates with intense red fluorescence. On the other hand, in apoptotic cells (especially mitochondria-mediated apoptotic cells) with low ΔΨm, JC-1 remains in the monomeric form, which shows only green fluorescence. In our study, apigenin did not induce a low mitochondrial transmembrane potential (ΔΨm), maintaining red fluorescence from 99.93 % (DMSO; MCF-7 vec) to 99.57 % (apigenin 40 μM; MCF-7 vec) and from 99.88 % (DMSO; MCF-7 HER2) to 97.56 % (apigenin 40 μM; MCF-7 HER2) as compared to FCCP (positive control, from 99.93 % to 92.89 % in MCF-7 vec and from 99.88 to 92.34 % in MCF-7 HER2) (Fig. 4a, b). We also measured the levels of BCL2 family members (BAX and BCL2) which are important in intrinsic mitochondrial apoptosis pathway. We found that apigenin failed to regulate the levels of BCL-XL, BAX or BCL2 in both cell types as seen in Fig. 4c. These results demonstrate that apigenin does not induce apoptosis via the intrinsic mitochondrial apoptosis pathway in MCF-7 vec and MCF-7 HER2 breast cancer cells.


Apigenin does not induce apoptosis via the intrinsic mitochondrial apoptosis pathway in MCF-7 vec and MCF-7 HER2 cells. a, b MCF-7 vec and MCF-7 HER2 cells were incubated with apigenin (0–40 μM) for 72 h and were dyed with JC-1 (4 μg/ml). The data were analyzed by FACSCalibur flow cytometry measuring the green fluorescence and red fluorescence at 514/529 nm (FL-1) and 585/590 nm (FL-2), respectively. c Analysis of intrinsic apoptosis-related molecules. MCF-7 vec and MCF-7 HER2 cells were treated with apigenin (0–40 μM) for 24 h. Total proteins were analyzed by western blotting with anti-BCL2, -BAX -BCL-XL and -tubulin antibodies. The relative band intensities are indicated below each western blot picture
Apigenin induces apoptosis via the extrinsic apoptosis pathway in MCF-7 vec and MCF-7 HER2 breast cancer cells
In this step, we investigated whether apigenin activated the extrinsic apoptosis pathway by measuring the expression of death receptor signaling-related proteins including FAS, caspase-8, and PARP. We observed that apigenin up-regulated the level of cleaved caspase-8 and induced the cleavage of PARP in MCF-7 vec and MCF-7 HER2 cells (Fig. 5a). We did not detect caspase-3 which is important in the extrinsic apoptosis pathway since MCF-7 cells do not contain caspase-3 due to a genomic deletion [39]. This suggests that MCF-7 cells induce the cleavage of PARP via caspase-3-independent pathway. Our results indicate that apigenin induces apoptosis via extrinsic caspase-8 and PARP-dependent apoptosis pathway in MCF-7 vec and MCF-7 HER2 cells.

a Apigenin induces apoptosis via death receptor-mediated extrinsic apoptosis pathway in MCF-7 vec and MCF-7 HER2 cells. Analysis of intrinsic apoptosis-related molecules. MCF-7 vec and MCF-7 HER2 cells were treated with apigenin (0–40 μM) for 24 h. Whole-cell lysates were analyzed by western blotting with anti-FAS, -cleaved caspase-8, -PARP and -tubulin antibodies. The relative band intensities are indicated below each western blot picture. b Apigenin increases the expression of p53, p-p53, and p21 in MCF-7 vec and MCF-7 HER2 cells. MCF-7 vec and MCF-7 HER2 cells were treated with apigenin (0–40 μM) for 24 h. Whole-cell lysates were analyzed by western blotting with anti-p-HER2, -HER2, -p53, -p21, -p-p53, -p-MDM2, -MDM2, and -tubulin antibodies. The relative band intensities are indicated below each western blot picture
Apigenin increases the expression of p53, phospho-p53 and p21 in MCF-7 vec and MCF-7 HER2 breast cancer cells
We next measured the expression of protein related to apoptosis by western blot analysis. As seen in Fig. 5b, apigenin (40 μM) reduced the tyrosine phosphorylation of HER2 (phospho-HER2 level) in MCF-7 HER2 cells. On the other hand, apigenin increased the levels of p53, phospho-p53 (p-p53), and p21 (p53 target gene) in MCF-7 vec and MCF-7 HER2 cells suggesting that apigenin displays antiproliferative activity via p53-dependent pathway. It is known that p53 is negatively regulated by the E3 ubiquitin ligase MDM2. We found that p-MDM2 expression was slightly reduced in both MCF-7 vec and MCF-7 HER2 cells under apigenin treatment (40 μM).
Apigenin failed to reduce the expression of AKT, ERK, JNK, and p38
Next, we investigated whether apigenin reduces intracellular signaling molecules such as AKT, ERK, JNK, and p38 which are implicated in cell proliferation and survival. As seen in Fig. 6a, western blot analysis demonstrated that apigenin failed to reduce the level of AKT, ERK, JNK, p38 and their active forms (phospho forms) in MCF-7 vec and MCF-7 HER2 cells.

a Effect of apigenin on the expression of intracellular signaling molecules in MCF-7 vec and MCF-7 HER2 cells. MCF-7 vec and MCF-7 HER2 cells were treated with apigenin (0–40 μM) for 24 h. Whole-cell lysates were analyzed by western blotting for the detection of specific protein, as indicated (p-AKT, AKT, p-ERK, ERK, p-JNK, JNK, p-p38, p38, and tubulin). The relative band intensities are indicated below each western blot picture. b Effect of apigenin on the expression of STAT3 in MCF-7 vec and MCF-7 HER2 cells. MCF-7 vec and MCF-7 HER2 cells were treated with apigenin (0–40 μM) for 24 h. Whole-cell lysates were analyzed by western blotting for the detection of specific protein, as indicated (p-JAK1, p-STAT3, STAT3, and tubulin). The relative band intensities are indicated below each western blot picture. c Effect of apigenin on the STAT3 transcriptional activity in MCF-7 vec and MCF-7 HER2 cells. Hep3B-pSTAT3-luciferase cells were treated with 40 μM apigenin and/or IL-6 (10 ng/ml) for 24 h. Cell lysates were submitted to promega luciferase assay
Apigenin inhibits the STAT3 signaling in MCF-7 vec and MCF-7 HER2 breast cancer cells
As seen Fig. 6b, apigenin reduced the expression of active form of signal transducer and activator of transcription 3 (p-STAT3) as well as p-JAK1 (upstream kinase of STAT3) in both MCF-7 vec and MCF-7 HER2 cells. Moreover, apigenin decreased STAT3 transcriptional activity as revealed by STAT3-dependent luciferase reporter gene assay in Hep3B-pSTAT3-luciferase stable cell line (Fig. 6c). This suggests that apigenin decreased cell growth rate inhibiting STAT3 signaling pathway.
Apigenin inhibits the NFκB signaling in MCF-7 vec and MCF-7 HER2 breast cancer cells
To investigate whether apigenin demonstrates its antiproliferative activity by inhibition of NFκB signaling pathway, we measured the NFκB signaling molecules by western blot analysis. As seen in Fig. 7a, we found that apigenin inhibited the phosphorylation of IκBα in cytosol extracts, suggesting that it abrogates the dissociation of IκBα from NFκB heterodimer (p65 and p50) blocking the proteasomal degradation of IκBα. Apigenin also inhibited the translocation of p65 (subunit of NFκB heterodimer) within the nucleus, leading to the inhibition of the transactivation of NFκB target genes (Fig. 7a). These immunoblotting data were confirmed by immunocytochemical result. As seen in Fig. 7b, 40 μM apigenin reduced nuclear staining intensity of NFκB as compared to control (DMSO treatment).

Effect of apigenin on NFκB signaling pathway in MCF-7 vec and MCF-7 HER2 cells. MCF-7 vec and MCF-7 HER2 cells were treated with 40 μM apigenin for 24 h. a Nuclear and cytosol extract were prepared as indicated in “Methods and materials” and were analyzed by western blotting for the detection of specific protein, as indicated (p65, Lamin B, p-IκBα, IκBα, and tubulin). The relative band intensities are indicated below each western blot picture. b Cells were submitted to the immunocytochemistry for the detection of p65
Discussion
In this study, we investigated the antiproliferative activity of apigenin and its mechanism of action in HER2-overexpressing breast cancer cells. Apigenin suppressed the growth of MCF-7 vec and MCF-7 HER2 cells (MCF-7 engineered to overexpress HER2 oncogene, ER-positive) in a dose-dependent manner. We did not find any resistance of HER2 to apigenin for the growth inhibition. Apigenin also inhibited the growth of SK-BR-3 cells (ER-negative) which express naturally HER2 in a dose-dependent manner. It should be noted that the intensity of cell growth inhibition induced by apigenin was stronger in MCF-7 cells as compared to SK-BR-3 cells. Moreover, we investigated the implication of ER in the pro-apoptotic action of apigenin. These are the reason why we chose MCF-7 cells engineered to overexpress HER2 for our experiments. Previously, we reported that apigenin induces growth suppression and apoptosis in MDA-MB-231 cells (ER-negative) with increase of p-p53 and p21 suggesting that it acts via ER-independent manner [26]. In this study, we found that apigenin induces growth suppression and apoptosis in MCF-7 vec and MCF-7 HER2 cells. Hence, it seems that apigenin induces apoptosis by both ER-dependent and -independent manner.
Apigenin is known to reduce total HER2 protein expression and induce apoptosis in HER2 overexpressing cells [40]. In our system, apigenin inhibited the tyrosine phosphorylation of HER2 in MCF-7 HER2 cells. Interestingly, apigenin increased the expression of p53, active p53 (p-p53) and p21 (p53 target gene), suggesting that this compound suppresses HER2-overexpressing cancer cell growth via a p53-dependent manner. In agreement with our data, apigenin has been shown to induce G1 or G2/M arrest and apoptosis in human prostate carcinoma cells [22, 23], human cervical carcinoma cells [41], and human hepatoma cells [42] through p53-dependent manner. The p53 tumor suppressor inhibits cellular proliferation by inducing cell cycle arrest and apoptosis in response to cellular stresses including DNA damage, growth factor deprivation, hypoxia, and oncogene activation [43, 44]. p53-dependent apoptosis is produced by the caspase proteinases and related to pro-apoptotic proteins such as BAX, NOXA, and PUMA [43]. p53-independent apoptosis is associated with the presence of p73 and the transcription factor E2F-1 [45]. p53 and E2F-1 are partners for apoptosis [46]. p53 induces growth arrest and cell death via negative regulation of MDM2 (MDM2 inhibits the tumor suppressive activity of p53) and positive regulation of ARF (ARF inhibits MDM2 activity) (ARF-MDM2-p53 pathway). In the INK4A-Rb-E2F pathway, Rb suppresses E2F activity and INK4A helps Rb activity, inhibiting cyclin D, CDK4, or CDK6 to induce apoptosis. ARF-MDM2-p53 pathway and INK4A-Rb-E2F pathway cross talk as reported elsewhere [46]. We found that apigenin slightly reduced p-MDM2 in MCF-7 vec and MCF-7 HER2 cells suggesting ARF-MDM2-p53 apoptosis pathway in our system.
The growth inhibition by apigenin was related to the increase of the sub G0/G1 apoptotic fraction in MCF-7 vec and MCF-7 HER2 cells. Interestingly, apigenin did not induce apoptosis via the intrinsic mitochondrial apoptosis pathway since this compound did not reduce the mitochondrial membrane potential, maintaining red fluorescence, and did not affect the levels of BCL2 and BAX. BCL2 family includes both anti-apoptotic protein (BCL2, BCL-XL, MCL-1, and CED-9, etc.) and pro-apoptotic protein (BAX, BAK, DIVA, BCL-Xs, BIK, and BIM, etc.) [47]. All BCL2 members possess at least one of four conserved motifs known as BCL2 homology domains (BH1–BH4) [47]. Pro- and anti-apoptotic family members can heterodimerize and regulate each other’s functions; their relative concentration may be important in modulating apoptosis [47]. On the other hand, apigenin induced the extrinsic apoptosis pathway: apigenin activated death receptor-related apoptosis signaling, enhancing the cleavage of caspase-8 and PARP.
Interestingly, apigenin was unable to inhibit the cell survival signaling such as AKT, ERK, JNK, and p38. However, apigenin reduced the level of p-STAT3 and p-JAK1 and decreased STAT3 transcriptional activity in MCF-7 vec and MCF-7 HER2 cells indicating that this compound induces growth-suppressive activity inhibiting STAT3-signaling pathway. STAT3 is a transcription factor and regulates the expression of various genes in response to cell stimuli and plays an important role in cell growth and apoptosis. STAT3 acts usually as a tumor promoter, although its tumor suppressor role is recently reported [48, 49]. STAT3 accelerates cell proliferation and angiogenesis, inhibit apoptosis, and drive invasion and metastasis [50–52]. STAT3 in melanoma tumors is associated with poor prognosis [50–52]. Constitutive STAT3 phosphorylation is mediated by several upstream kinases (JAK and Src) and is thought to be a key component of the oncogenic process [53, 54]. Phytoestrogen (resveratrol) is known to inhibit STAT3 signaling and induces the apoptosis of malignant cells containing activated STAT3 [55].
NFκB is a transcription factor and plays an important role in cell proliferation, survival, inflammation, immune response, tumor formation, and tumor progression [56, 57]. The NFκB family consists of p50/p105 (NFκB1), p52/p100 (NFκB2), REL A (p65), c-REL and REL B [56, 57]. In cells, the p65–p50 heterodimer is maintained in an inactive state due to binding of IκB. However, when IκB is phosphorylated via IκB kinase, phospho-IκB is detached from p65 to p50 dimer to be degraded. Free p65–p50 heterodimer then can enter the nucleus, and bind to specific DNA sequences (κB site) to induce the transcription of target genes related to tumor promotion, cell survival signaling, and inflammation [56, 57]. In our study, apigenin suppressed the growth of MCF-7 vec and MCF-7 HER2 cells, inhibiting the NFκB signaling pathway. Clearly, apigenin abrogated the phosphorylation of IκBα, and nuclear translocation of p65 within the nucleus.
About 20–25 % of invasive breast carcinomas have HER2 gene amplification [58]. A normal breast cell has 20,000 HER2 receptors but a breast cancer cell could have 1.5 million. HER2 is a member of HER/ErbB2/Neu protein, which also includes HER1/EGFR, HER3, and HER4. HER2 cross talks with ER signal transduction pathway [59] and its expression level can be regulated by ER. In our study, we found that apigenin significantly inhibited the growth of MCF-7 HER2-overexpressing cancer cells. This indicates that apigenin could be a useful natural therapy that inhibits HER2-overexpressing breast cancer. Apigenin could be promising products to treat and prevent HER2-overexpressing breast cancer.
References
Zhang A, Wu Y, Lai HWL, Yew DT (2004) Apoptosis—a brief review. Neuroembryology 3:47–59
Kolb TM, Chang SH, Davis MA (2002) Biochemical and morphological events during okadaic acid-induced apoptosis of Tsc2-null ERC-18 cell line. Toxicol Pathol 30:235–246
Waring P, Mullbacher A (1999) Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol 77:312–317
Gupta S (2003) Molecular signaling in death receptor and mitochondrial pathways of apoptosis (review). Int J Oncol 22:15–20
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274:22932–22940
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
Robinson AJ, Kunji ER, Gross A (2012) Mitochondrial carrier homolog 2 (MTCH2): the recruitment and evolution of a mitochondrial carrier protein to a critical player in apoptosis. Exp Cell Res. doi:10.1016/j.yexcr.2012.01.026
Qin S, Yang C, Li S, Xu C, Zhao Y, Ren H (2012) Smac: its role in apoptosis induction and use in lung cancer diagnosis and treatment. Cancer Lett 318:9–13
Gartel AL (2012) Mechanisms of apoptosis induced by anticancer compounds in melanoma cells. Curr Top Med Chem 12:50–52
Sirtori CR, Arnoldi A, Johnson SK (2005) Phytoestrogens: end of a tale? Ann Med 37:423–438
Mense SM, Hei TK, Ganju RK, Bhat HK (2008) Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ Health Perspect 116:426–433
Velentzis LS, Woodside JV, Cantwell MM, Leathem AJ, Keshtgar MR (2008) Do phytoestrogens reduce the risk of breast cancer and breast cancer recurrence? What clinicians need to know. Eur J Cancer 44:1799–1806
Duffy C, Cyr M (2003) Phytoestrogens: potential benefits and implications for breast cancer survivors. J Womens Health (Larchmt) 12:617–631
Duffy C, Perez K, Partridge A (2007) Implications of phytoestrogen intake for breast cancer. CA Cancer J Clin 57:260–277
Zhou JR, Gugger ET, Tanaka T, Guo Y, Blackburn GL, Clinton SK (1999) Soybean phytochemicals inhibit the growth of transplantable human prostate carcinoma and tumor angiogenesis in mice. J Nutr 129:1628–1635
de Souza PL, Russell PJ, Kearsley JH, Howes LG (2010) Clinical pharmacology of isoflavones and its relevance for potential prevention of prostate cancer. Nutr Rev 68:542–555
Toyomura K, Kono S (2002) Soybeans, soy foods, isoflavones and risk of colorectal cancer: a review of experimental and epidemiological data. Asian Pac J Cancer Prev 3:125–132
Singh AV, Franke AA, Blackburn GL, Zhou JR (2006) Soy phytochemicals prevent orthotopic growth and metastasis of bladder cancer in mice by alterations of cancer cell proliferation and apoptosis and tumor angiogenesis. Cancer Res 66:1851–1858
Adjakly M, Bosviel R, Rabiau N, Boiteux JP, Bignon YJ, Guy L, Bernard-Gallon D (2011) DNA methylation and soy phytoestrogens: quantitative study in DU-145 and PC-3 human prostate cancer cell lines. Epigenomics 3:795–803
Qi W, Weber CR, Wasland K, Savkovic SD (2011) Genistein inhibits proliferation of colon cancer cells by attenuating a negative effect of epidermal growth factor on tumor suppressor FOXO3 activity. BMC Cancer 11:219
Gupta S, Afaq F, Mukhtar H (2002) Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 21:3727–3738
Shukla S, Gupta S (2006) Molecular targets for apigenin-induced cell cycle arrest and apoptosis in prostate cancer cell xenograft. Mol Cancer Ther 5:843–852
Liang YC, Huang YT, Tsai SH, Lin-Shiau SY, Chen CF, Lin JK (1999) Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis 20:1945–1952
Birt DF, Mitchell D, Gold B, Pour P, Pinch HC (1997) Inhibition of ultraviolet light induced skin carcinogenesis in SKH-1 mice by apigenin, a plant flavonoid. Anticancer Res 17:85–91
Seo HS, Ju JH, Jang K, Shin I (2011) Induction of apoptotic cell death by phytoestrogens by up-regulating the levels of phospho-p53 and p21 in normal and malignant estrogen receptor α-negative breast cells. Nutr Res 31:139–146
Lu HF, Chie YJ, Yang MS, Lee CS, Fu JJ, Yang JS, Tan TW, Wu SH, Ma YS, Ip SW, Chung JG (2010) Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax- and Bcl-2-triggered mitochondrial pathway. Int J Oncol 36:1477–1484
Wang W, Heideman L, Chung CS, Pelling JC, Koehler KJ, Birt DF (2000) Cell-cycle arrest at G2/M and growth inhibition by apigenin in human colon carcinoma cell lines. Mol Carcinog 28:102–110
Turktekin M, Konac E, Onen HI, Alp E, Yilmaz A, Menevse S (2011) Evaluation of the effects of the flavonoid apigenin on apoptotic pathway gene expression on the colon cancer cell line (HT29). J Med Food 14:1107–1117
Ruela-de-Sousa RR, Fuhler GM, Blom N, Ferreira CV, Aoyama H, Peppelenbosch MP (2010) Cytotoxicity of apigenin on leukemia cell lines: implications for prevention and therapy. Cell Death Dis 1:e19
Ujiki MB, Ding XZ, Salabat MR, Bentrem DJ, Golkar L, Milam B, Talamonti MS, Bell RH Jr, Iwamura T, Adrian TE (2006) Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer 5:76
Spevak CC, Park EH, Geballe AP, Pelletier J, Sachs MS (2006) Her-2 upstream open reading frame effects on the use of downstream initiation codons. Biochem Biophys Res Commun 350:834–841
Nielsen DL, Andersson M, Kamby C (2009) HER2-targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev 35:121–136
Ménard S, Pupa SM, Campiglio M, Tagliabue E (2003) Biologic and therapeutic role of HER2 in cancer. Oncogene 22:6570–6578
Lipton A, Ali SM, Leitzel K, Demers L, Chinchilli V, Engle L, Harvey HA, Brady C, Nalin CM, Dugan M, Carney W, Allard J (2002) Elevated serum her-2/neu level predicts decreased response to hormone therapy in metastatic breast cancer. J Clin Oncol 20:1467–1472
Tokunaga E, Oki E, Nishida K, Koga T, Egashira A, Morita M, Kakeji Y, Maehara Y (2006) Trastuzumab and breast cancer: developments and current status. Int J Clin Oncol 11:199–208
Dean-Colomb W, Esteva FJ (2008) Her2-positive breast cancer: herceptin and beyond. Eur J Cancer 44:2806–2812
Shin I, Arteaga CL (2006) Expression of active akt protects against tamoxifen-induced apoptosis in MCF-7 cells. IUBMB Life 58:664–669
Jänicke RU (2009) MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat 117:219–221
Way TD, Kao MC, Lin JK (2004) Apigenin induces apoptosis through proteasomal degradation of HER2/neu in HER2/neu-overexpressing breast cancer cells via the phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem 279:4479–4489
Zheng PW, Chiang LC, Lin CC (2005) Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci 76:1367–1379
Chiang LC, Ng LT, Lin IC, Kuo PL, Lin CC (2006) Anti-proliferative effect of apigenin and its apoptotic induction in human Hep G2 cells. Cancer Lett 237:207–214
Schuler M, Green DR (2001) Mechanisms of p53-dependent apoptosis. Biochem Soc Trans 29:684–688
Shen Y, White E (2001) p53-dependent apoptosis pathways. Adv Cancer Res 82:55–84
Stiewe T, Putzer BM (2000) Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 26:464–469
Polager S, Ginsberg D (2009) p53 and E2F: partners in life and death. Nat Rev Cancer 9:738–748
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A (2008) Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Gene Dev 22:449–462
Lewis HD, Winter A, Murphy TF, Tripathi S, Pandey VN, Barton BE (2008) STAT3 inhibition in prostate and pancreatic cancer lines by STAT3 binding sequence oligonucleotides: differential activity between 5′ and 3′ ends. Mol Cancer Ther 7:1543–1550
Kortylewski M, Jove R, Yu H (2005) Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev 24:315–327
Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, Chang A, Kraker A, Jove R, Yu H (2002) Roles of activated Src and STAT3 signaling in melanoma tumor cell growth. Oncogene 21:7001–7010
Xie TX, Huang FJ, Aldape KD, Kang SH, Liu M, Gershenwald JE, Xie K, Sawaya R, Huang S (2006) Activation of STAT3 in human melanoma promotes brain metastasis. Cancer Res 66:3188–3196
Sellers LA, Feniuk W, Humphrey PP, Lauder H (1999) Activated G protein-coupled receptor induces tyrosine phosphorylation of STAT3 and agonist-selective serine phosphorylation via sustained stimulation of mitogen-activated protein kinase. Resultant effects on cell proliferation. J Biol Chem 274:16423–16430
Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, Krolewski JJ, Medveczky P, Jove R (2000) Activation of STAT3 in v-Src-transformed fibroblasts requires cooperation of JAK1 kinase activity. J Biol Chem 275:24935–24944
Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R (2006) Resveratrol inhibits Src and STAT3 signaling and induces the apoptosis of malignant cells containing activated STAT3 protein. Mol Cancer Ther 5:621–629
He G, Karin M (2011) NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res 21:159–168
Xiao G, Fu J (2011) NF-κB and cancer: a paradigm of Yin-Yang. Am J Cancer Res 1:192–221
Tolaney SM, Krop IE (2009) Mechanisms of trastuzumab resistance in breast cancer. Anticancer Agents Med Chem 9:348–355
Buzdar AU (2009) Role of biologic therapy and chemotherapy in hormone receptor- and HER2-positive breast cancer. Ann Oncol 20:993–999
Acknowledgments
This study was supported by a Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0063466). This study was also supported by a grant of the Traditional Korean Medicine R&D Project, Ministry of Health & Welfare, Republic of Korea (B110043).
Conflict of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seo, HS., Choi, HS., Kim, SR. et al. Apigenin induces apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3 and NFκB signaling in HER2-overexpressing breast cancer cells. Mol Cell Biochem 366, 319–334 (2012). https://doi.org/10.1007/s11010-012-1310-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1310-2