
Abstract
Cyclophosphamide (CPM), an alkylating agent is used as an immunosuppressant in rheumatoid arthritis and in the treatment of several cancers as well. In this study, Ellagic acid (EA), a naturally occurring plant polyphenol, was evaluated for its antigenotoxicity and antioxidant efficacy against the CPM-induced renal oxidative stress and genotoxicity in Swiss albino mice. The mice were given a prophylactic treatment of EA orally at a dose of 50 and 100 mg/kg body weight (b wt) for seven consecutive days before the administration of a single intraperitoneal (i.p.) injection of CPM at 50 mg/kg b wt. The modulatory effects of EA on CPM-induced nephrotoxicity and genotoxicity were investigated by assaying oxidative stress biomarkers, serum kidney toxicity markers, DNA fragmentation, alkaline unwinding assay, micronuclei (MN) assay, and by histopathological examination of kidney tissue. A single intraperitoneal administration of CPM in mice increased malondialdehyde level with depletion in glutathione content, antioxidant enzymes activities, viz. glutathione peroxidase, glutathione reductase, catalase, quinone reductase, induced DNA strand breaks, and MN induction. EA oral administration at both doses caused significant reduction in their levels, restoration in the activities of antioxidant enzymes, reduction in MN formation, and DNA fragmentation. Serum toxicity marker enzymes like BUN, creatinine, and LDH were also increased after CPM treatment which was significantly decreased in EA pretreated groups. Present findings suggest a prominent role of EA against CPM-induced renal injury, DNA damage, and genotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural products have been shown to be an excellent and reliable source for the development of new drugs [1]. Epidemiologic studies have showed that there is an association between the intake of phenolic foods and the protection from various diseases [2]. These phenolic compounds have tremendous antioxidant and chemoprotective properties in vivo [3]. Phenolic compounds are the most promising anticarcinogenic agents in plants [4]. Ellagic acid (EA) has been reported to inhibit peroxynitrite-induced oxidation and nitration reactions [5]. EA is a potent dietary antioxidant found in variety of fruits, nuts, and many other food sources. EA is reported to exhibit antioxidant, antiproliferative, radical scavenging, antiapoptotic, antiatherogenic, antiasthmatic, estrogen receptor modulator, and chemopreventive, in a variety of tissues and cells. It has been also shown to inhibit cancer caused by several types of chemical carcinogens including polycyclic aromatic hydrocarbons, N-nitrosamines, aflatoxin B1, and aromatic amines [6–15]. EA also finds application in cosmetology as a skin whitening agent as it inhibits radiation induced melanogenesis. EA contains four hydroxyl groups and two lactone groups in which hydroxyl group is known to increase antioxidant activity in lipid peroxidation and protect cells from oxidative damage [16] and few more reported that EA decrease hepatic cytochrome P450 activity and also increase the activities of several phase II enzymes [17–19].
Cyclophosphamide (CPM) is a well-known bi-functional alkylating agent widely used in cancer chemotherapy and expresses its genotoxicity when metabolically activated [20, 21]. It is extensively used for the treatment of various cancers as well as an immunosuppressant in organ transplantation, rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, and other benign diseases [22, 23]. According to the International Agency for Research on Cancer (IARC), CPM is widely used as reference mutagen and has been classified as carcinogenic for animals and humans [24]. Reactive metabolites of CPM chemically alkylate DNA as well as protein, producing cross-links which are responsible for its cytotoxic effect [25]. Normal tissues injury or damage is the major limitation of using CPM, which gives rise to numerous side effects, CPM treatment also results in the production of reactive oxygen species (ROS), which cause peroxidative damage to kidney and other vital organs [26]. Antineoplastic and toxic effects like necrosis, apoptosis, and oncosis of CPM are linked with two active metabolites, i.e., phosphoramide and acrolein. Further Tripathi and Jena [27] reported CPM to be toxic in germ cells of mice, it has also been reported that CPM treatment for non-Hodgkin’s lymphoma leads to the induction of secondary cancers in bladder and kidney [28]. The important factor for therapeutic and toxic effects of CPM is the requirement of metabolic activation by hepatic microsomal cytochrome P450 mixed functional oxidase system [29, 30]. On activation, CPM generates active alkylating metabolites such as 4-hydroxycyclophosphamide, acrolein, and aldophosphamide mustard, which hamper with cellular DNA synthesis in fast dividing cells and ultimately lead to cell death [31]. Bagley and Bostick [32] have reviewed the toxicity and pharmacology of CPM.
In this study, we have made an attempt to investigate the beneficial effects of EA against CPM-induced nephrotoxicity and genotoxicity. The experimental end points for nephrotoxicity included biochemical estimation of antioxidant enzymes and histopathological measurement for the determination of oxidative stress. Genotoxicity end points included evaluation of micronuclei (MN), DNA fragmentation, and alkaline unwinding assay in terms of DNA strand breaks. Our results reveal that EA decreased the CPM-induced oxidative stress and subsequent oxidative DNA damage and genotoxicity in Swiss albino mice.
Materials and methods
Chemicals
EA, EDTA, Tris, reduced glutathione (GSH), oxidized glutathione, nicotinamide adenine dinucleotide phosphate reduced (NADPH), CPM, bovine serum albumin, bisbenzamide; Proteinase K, EDTA, SDS, phenol, chloroform, isoamyl alcohol and RNase were from Sigma St. Louis. All other reagents and solvents were of a high analytical grade.
Animals
Eight-week-old adult male Swiss albino mice (20–25 g) were obtained from the Central Animal House Facility of Hamdard University, New Delhi and were housed in a ventilated room at 25 ± 5°C under a 12-h light/dark cycle. The animals were acclimatized for 1 week before the study and had free access to standard laboratory feed (Hindustan Lever Ltd., Bombay, India) and water ad libitum. The study was approved by the Committee for the Purpose of Control and Supervision of Experimental Animals.
Experimental design
For study of biochemical parameters and MN assay, 25 male Swiss albino mice were divided into five groups. Group I served as a saline-treated control. Group II served as toxicant and was given single intraperitoneal injection of CPM (50 mg/kg b wt i.p.) freshly dissolved in distilled water. EA was orally given at two doses, 50 and 100 mg/kg b wt, [33, 34] to groups III and IV, respectively, for seven consecutive days. Group V received only a high dose of EA for seven consecutive days. On the seventh day (last day) of pretreatment, single intraperitoneal dose of CPM (50 mg/kg b wt i.p.) was administered to the animals in groups II, III, and IV. All the animals were killed after 24 h of intoxication with CPM. Kidneys and femur bones were removed and processed for enzyme estimation and MN assay. The dose of CPM was selected on the basis of previous reports from our lab and others [35–37].
Micronucleus assay
The micronucleus test (mouse bone marrow) was performed according to the method of Schmid [38]. In brief, the air-dried slides were stained with May–Grunwald and Giemsa as described by Schmid, made permanent, and coded. A total no. of 1,500–2,000 Normochromatic erythrocytes (NCE) and polychromatic erythrocytes (PCEs) were scored per animal by the same viewer for determining the frequencies of micronucleated PCE (MnPCEs). To detect possible cytotoxic effects, the PCE:NCE ratio in 200 erythrocytes per animal was calculated according to Gollapudi and McFadden [39]. Coded slides were scored with a light microscope at a ×1000 magnification.
DNA isolation
DNA extraction was done by standard chloroform isoamyl method. DNA was extracted from approximately 400 mg of kidney tissue by homogenizing the tissue in 5 mL TNE buffer (50 mM Trisma, 100 mM EDTA, 0.5% SDS, pH 8.0) in a 2-mL ground glass homogenizer. Each sample was homogenized with ten standardized strokes of the pestle to minimize any potential effect on DNA integrity introduced by the homogenization procedure. An equal volume of buffered phenol/chloroform/isoamyl alcohol (PCI) (25:24:1, v/v/v, pH 8.0) was then added to the sample. The sample was gently mixed and allowed to settle for 5 min and then centrifuged for 5 min at 13,000 rpm at 4°C. The aqueous layer was transferred to a new micro centrifuge tube and PCI extraction was repeated. The aqueous layer was then digested by 5 mL of RNase (10 mg/mL) for 30 min at 37°C and the digest was extracted once by PCI and once by 500 mL of chloroform. DNA was precipitated from the resulting aqueous layer by adding two volumes of absolute ethanol and 1/10 volume of 3 M sodium acetate, pH 5.2. The sample was then centrifuged (13,000 rpm, 15 min), and the resulting pellet rinsed with 500 mL of 70% ethanol and air-dried. The amount of DNA was quantitated spectrophotometrically at 260 and 280 nm. 2 mg/mL of DNA sample was dissolved in 1 mL of TE buffer (10 mM Trisma, 1 mM EDTA) and subsequently used in the DNA alkaline unwinding assay.
Alkaline unwinding assay
In the alkaline unwinding assay, the rate of transition of double stranded DNA (dsDNA) to single stranded DNA (ssDNA) under predefined alkaline denaturing condition was proportional to the number of breaks in the phosphodiester backbone and thus was used as a measure of DNA integrity. Bisbenzamide was used as a DNA-binding dye from the fluorescence of which various types of DNA were quantitated. For the fluorescence determination of dsDNA, ssDNA, and partially unwound DNA (au-DNA), three equal portions of diluted DNA sample was prepared. The amount of dsDNA was obtained from the fluorescence of a sample without any treatment; while ssDNA was determined from the sample that had been boiled for 30 min. Fluorescence of the DNA sample which had been subjected to alkaline treatment (pH 12.2) on ice for 30 min provided an estimate of the amount of au-DNA. The fluorescence of initial or dsDNA was determined by placing 100 μmol DNA sample, 100 μL NaCl (25 mM) and 2 μL SDS (0.5%) in a prechilled test tube, followed by the addition of 3 mL 0.2 M potassium phosphate pH 9, and 3 μL bisbenzamide (1 mg/mL). The contents were mixed and allowed to react in the dark for 15 min to allow the fluorescence to stabilize. The fluorescence of the sample was the measured using a spectrofluorimeter (Ex: 360 nm, Em: 450 nm). The fluorescence of ssDNA was determined as above but using the DNA sample that had already been boiled for 30 min to completely unwind the DNA. 50 μL NaOH (0.05 N) was rapidly mixed with 100 μL of DNA sample in a prechilled test tube. The mixture was incubated on ice in the dark for 30 min followed by rapid addition and mixing of 50 μL HCl (0.05 N). This was followed immediately by an addition of 2 μL SDS (0.5%) and the mixture was forcefully passed through a 21 G needle five times. Fluorescence of alkaline unwound DNA sample was measured as described above. Measurement of the alkaline unwound sample was performed in triplicate and the average was reported. The ratio between dsDNA to total DNA (F value) was determined as follows:
where au-DNA, ssDNA, and dsDNA were the degrees of fluorescence from the partially unwound, single stranded, and double stranded determinations, respectively. The F value was inversely proportion to the number of strand breaks present and thus could be used as an indicator of DNA integrity.
Gel electrophoresis and DNA fragmentation (smearing)
The sample was mixed with 10 μL of loading solution (10 mM EDTA (pH 8.0), 1% (w/v) bromophenol blue, and 40% (w/v) sucrose) preheated to 70°C. The DNA samples were loaded onto a 1.8% (w/v) agarose gel and sealed with 0.8% (w/v) low melting point agarose. The DNA fragments were separated by electrophoresis at 25 V for 12 h at 4°C in TBE buffer. The DNA was visualized using ethidium bromide and photographed by digital camera.
Postmitochondrial supernatant preparation
Kidneys were removed quickly, cleaned free of extraneous material, and immediately perfused with ice-cold saline (0.85% sodium chloride). The kidneys were homogenized in chilled phosphate buffer (0.1 M, pH 7.4) containing potassium chloride (KCl; 1.17%) using a Potter Elvehjem homogenizer. The homogenate was filtered through muslin cloth and was centrifuged at 800×g for 5 min at 4°C by REMI Cooling Centrifuge to separate the nuclear debris. The aliquot so obtained was centrifuged at 12,000 rpm for 20 min at 4°C to obtain postmitochondrial supernatant which was used as a source of enzymes. All the biochemical estimations were completed within 24 h of killing the animal [40].
GSH estimation was done by the method of Jollow et al. [41].
Glutathione reductase (GR) activity was estimate by the method of Carlberg and Mannervik [42].
Glutathione peroxidase (GPx) activity was performed by Mohandas et al. [43].
Glutathione-S-transferase activity was estimated by Habig et al. [44].
Malondialdehyde (MDA) formation was estimated by Wright et al. [45].
Xanthine oxidase (XO) estimation was done by Stripe and Della Corte [46].
Catalase activity was measured by Claiborne [47].
Lactate dehydrogenase (LDH) activity was measured by the method of Kornberg [48].
Estimation of protein concentration was done by Lowry et al. [49].
Estimation of blood urea nitrogen (BUN), γ-glutamyl transferase and creatinine was done by commercially available kit from Rekon diagnostics Pvt Limited.
Histopathological examination
The kidneys were quickly removed after killing the mice and preserved in 10% neutral buffered formalin for histopathological processing. The kidneys were embedded in paraffin wax and longitudinally sectioned under microtome. Hematoxylin and eosin staining of the above sections was done and observed under Olympus microscope.
Statistical analysis
Differences between groups were analyzed using analysis of variance (ANOVA) followed by Dunnet’s multiple comparisons test. All data points are presented as the treatment groups mean ± standard error of the mean (S.E.M).
Results
Effects of pretreatment of EA on renal glutathione, its dependent enzymes, and antioxidant enzyme system
Table 1 shows that CPM administration leads to significant depletion of renal GSH (p < 0.001). In addition, there was marked inhibition of GR enzyme by (p < 0.001), GST (p < 0.001), and GPx (p < 0.001) as compared to control as shown in Table 1. Pretreatment with EA augmented renal GSH content by both the doses (p < 0.05, p < 0.001), GR (p < 0.01), GST (p < 0.01, p < 0.001), GPx (p < 0.05, p < 0.001). Similarly CPM treatment as shown in Table 1 inhibited catalase which was increased by EA prophylactic treatment at both doses. No significant difference was found in the only D2 group compared with control in any of the parameters.
Table 1 also shows that CPM administration leads to the significant enhancement in the levels of XO, MDA, and GGT (p < 0.001) in comparison to control. Pretreatment with EA improved renal XO (p < 0.001), MDA (p < 0.05, p < 0.001) formation and GGT (p < 0.01, p < 0.001).
Effects of pretreatment of EA on renal toxicity marker enzymes
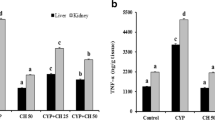
The effect of EA administration on CPM-mediated leakage of kidney toxicity markers (BUN, LDH, and creatinine) were shown in Figs. 1, 2, 3. Figures 1 and 2 showed that mice treated with CPM showed a significant increase in BUN (p < 0.001) and creatinine (p < 0.001) levels when compared with control. On treatment with EA marked inhibition was observed in BUN at both the doses (p < 0.001) (Fig. 1) and also in the creatinine level at both the doses (p < 0.01) (Fig. 2). Similar type of results was observed for LDH levels (Fig. 3). No significant difference was found in the only D2 group compared with control.

Results represent mean ± SE of six animals per group. Results obtained are significantly different from control group (***p < 0.001). Results obtained are significantly different from CPM-treated group (# p < 0.05), (## p < 0.01), and (### p < 0.001). EA Ellagic acid, D1 50 mg/kg b wt, D2 100 mg/kg b wt

Results represent mean ± SE of six animals per group. Results obtained are significantly different from control group (***p < 0.001). Results obtained are significantly different from CPM-treated group (# p < 0.05), (## p < 0.01), and (### p < 0.001). EA Ellagic acid, D1 50 mg/kg b wt, D2 100 mg/kg b wt

Results represent mean ± SE of six animals per group. Results obtained are significantly different from control group (***p < 0.001). Results obtained are significantly different from CPM-treated group (# p < 0.05), (## p < 0.01), and (### p < 0.001). EA Ellagic acid, D1 50 mg/kg b wt, D2 100 mg/kg b wt
Effects of EA pretreatment on genotoxicity
Table 2 shows that there was higher induction of MN in CPM-treated group (p < 0.001) and this level was decreased by prophylactic treatment of EA at low dose (p < 0.05) and more effectively at higher dose (p < 0.001). DNA damage is estimated in terms of smearing and lack of intact band control, only the EA treated groups at both doses showed less smearing and an intact band was also observed (Table 2). Similarly, in the DNA alkaline unwinding assay, a simultaneous significant decrease in F value (p < 0.001) on treatment with CPM was noted as compared to control group, whereas results indicate that there was significant increase in the F value at both the doses of EA (p < 0.05, p < 0.001).
The estimated PCE:NCE ratio in bone marrow preparations in the Table 2 shows a statistical decrease (p < 0.001) in hematopoiesis as a result of CPM treatment as compared to control. The pretreatment of EA at both the dose significantly increased the PCE:NCE ratio, indicating reversal of the cytotoxic effects caused by CPM administration to mice. In the Fig. 4, results indicate that there was significant DNA fragmentation only in the toxicant group as compared to the control group while there was less smearing in EA pretreated groups, EA pretreatment inhibited DNA disintegration.

Agarose (1.5%) gel electrophoresis of DNA obtained from mice kidney. Lane 1 control, Lane 2 CPM only, Lane 3 EA + CPM; D1, Lane 4 EA + CPM; D2, Lane 5 EA D2 only. From gel picture, it is marked that CPM treatment introduced DNA fragmentation by smearing of DNA when compared to control. There is decrease in DNA smearing as a result of EA pretreatment at both the doses in mice kidney. CPM cyclophosphamide, EA Ellagic acid, D1 50 mg/kg b wt, D2 100 mg/kg b wt
Histopathological examination
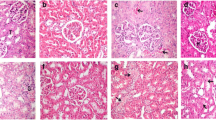
The control mice showed normal glomerular and tubular histology while CPM-administered mice were found to have necrosis and desquamation of epithelial cells, peritubular, and glomerular congestion and showed invasion of inflammatory cells in the cortical and medullary regions of kidneys. The nephrotoxicity was characterized by widespread degeneration of tubular architecture, tubular congestion, swelling and necrosis. In comparison, renal sections obtained from mice that were pretreated with EA at a dose of 100 mg/kg body weight demonstrated marked reduction of the histological character of renal injury. Prophylactic treatment of EA (50 mg/kg b wt) was also associated with a significant reduction in injury, but this reduction was less marked than in the higher dose EA pretreatment groups (Fig. 5).

Photomicrographs of kidney histology of Swiss albino mice (×400 magnifications). A Kidney section from control group shows normal renal histo-architecture. B Kidney section from only CPM-treated group shows loss of normal renal architecture, inflammatory cells infiltration, and fatty changes with cell swelling. C Kidney section of the EA pretreated groups at lower dose of EA (50 mg/kg b wt) moderately prevented the cytotoxic damage induced by CPM; as indicated by moderate swelling of the renal epithelial cells. D Kidney section of the EA pretreated groups at higher dose of EA (100 mg/kg b wt) improved the morphology of the kidney from the damage induced by CPM. E Higher dose of EA administration did not showed any kind of histological abnormalities in the kidney tissue. CPM cyclophosphamide, EA Ellagic acid, D1 50 mg/kg b wt, D2 100 mg/kg b wt, G glomeruli, T tubule, DCT distal convoluted tubule
Discussion
The kidney is the target of many xenobiotics toxicants, including drugs. There are many factors that contribute to the sensitivity of the kidney viz., presence of variety of metabolizing enzymes and xenobiotic transporters, large blood flow and concentration of solutes during urine production. Further physiological, anatomical, and biochemical features of the kidney make it particularly sensitive to many toxins and drugs. Present investigation was carried out with the aim of evaluating the possible role of EA in modulating the in vivo genotoxicity and oxidative renal injury of CPM.
The active metabolites of CPM are phosphoramide and acrolein, and they slow down the growth of cancer cells as they interfere with the cellular DNA. Hydroxycyclophosphamide and deschloroethylcyclophosphamide are the two intermediate compounds that lead to the formation of phosphoramide, the main agent responsible the mutagenic effect of CPM [4]. Phospharamide causes the induction of crosslink and strand lesion in DNA [50]. Studies have indicated that CPM has a pro-oxidant nature, and production of oxidative stress after CPM administration leads to decrease in the activities of antioxidant enzymes and increase in lipid peroxidation in different tissues of mice and rats [51, 52]. In our study also CPM treatment resulted in significant increases (p < 0.001) in the renal MDA with decrease in GSH (free thiols), GSH and its oxidized counterpart glutathione disulfide represent a major redox buffer system of the cell. GSH can act either as a non-enzymatic antioxidant by direct interaction of –SH group with ROS or it can be implicated in the enzymatic detoxification reaction for ROS, as a cofactor or coenzyme. The depletion of GSH content may be attributed to the direct conjugation of CPM and its metabolites with free or protein bound –SH groups and significant decreases (p < 0.001) in the GSH-Px and CAT activity in comparison to the control levels (Table 1). Moreover, Cooper et al. [53], Kehrer and Biswal [54] suggested that CPM treatment results in inflammation thus disturbing the overall renal redox cycling.CPM was also shown to cause decrease levels of other antioxidant enzymes like GR and glutathione-S-transferase. EA dose dependently increased the level of all the renal antioxidant enzymes to normal and this phenomenon was in agreement with the previously published reports [55, 56]. Simultaneously the EA pretreatment also augmented the depleted levels of GGT and XO in these mice.
The marked increase in the levels of serum BUN and creatinine is a marker for the nephrotoxicity and kidney damage [57]. CPM also induced renal damage characterized by increases in the level of LDH, BUN, and creatinine. CPM-induced elevations in the levels of LDH, BUN, and creatinine observed in our study are in agreement with the previously published reports by Cagler et al. [57]. The elevation in the activities of these enzymes in the serum might be due to the leakage of these cytosolic enzymes into the circulatory system resulting from kidney damage after CPM administration. This is indicative of the onset of renal damage due to kidney dysfunction and disturbance in the biosynthesis of these enzymes, with alteration in the membrane permeability. Administration with EA prevented CPM-induced renal toxicity, as indicated by a steep decrease in serum BUN, creatinine levels and LDH activity, possibly by maintaining the renal cellular membrane integrity. This is an indicator of possible nephro-protective efficacy offered by EA compared with the untreated and CPM-intoxicated groups.
Histopathological studies also provided supportive confirmation for the biochemical parameters depicted by the photomicrographs. Kidney tissue (Fig. 5) of CPM-administered mice shows tubular necrosis and desquamation of epithelial cells, invasion of inflammatory cells in the cortical and medullary regions of kidneys was also prominent. Lumen of tubules was observed to have collection of eosinophilic granular material. Another characteristic feature of CPM-induced toxicity is tubular renal epithelial cell hypertrophy which was observed in CPM-treated group and was in agreement with previously published report by Abrahama et al. [58]. The key histological finding of this study was that EA pretreatment influenced the recovery of kidney architecture induced by CPM.
Experimental evidences have strengthened the concept that, DNA damage plays a crucial role in the initiation and subsequent promotion of carcinogenesis induced by different genotoxicants. Errors in the DNA molecules cause chromosomal aberrations. MN are well characterized biomarkers of structural and numerical chromosomal damage. The MN in young erythrocytes arise mainly from chromosomal fragments that are not incorporated into the daughter nuclei at the time of cell division in the erythropoietic blast cells [59]. Decrease in the PCE:NCE ratio is responsible for the induction of bone marrow cytotoxicity [60]. Results of our study reveal that CPM treatment shows cytotoxicity and has highly clastogenic nature.CPM was found to induce MN formation as it decreased PCE:NCE ratio and EA effectively brought this PCE:NCE ratio to the normal level.CPM also results in the formation of DNA strand breaks as a result of interaction of acrolein with DNA and cause DNA disintegration or smearing due to necrosis and MnPCEs formation. Prophylactic treatment of EA significantly and dose dependently restores normal level all these hallmarks of genotoxicity.
This study shows the protective effect of EA against CPM-induced genotoxicity and nephrotoxicity. In conclusion, our study provides evidence that EA pretreatment attenuates CPM-induced oxidative stress and the subsequent DNA damage in mice. The antigenotoxic potential of EA might be due to its antioxidant property. Future investigation targeting molecular pathways involved in modulatory action of EA on CPM-induced genotoxicity and nephrotoxicity are needed.
Abbreviations
- EA:
-
Ellagic acid
- CPM:
-
Cyclophosphamide
- GSH:
-
Reduced glutathione
- GPx:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- XO:
-
Xanthine oxidase
- MDA:
-
Malondialdehyde
- BUN:
-
Blood urea nitrogen
- LDH:
-
Lactate dehydrogenase
References
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477
Morton LW, Abu-Amsha Caccetta R, Puddey IB, Croft KD (2000) Chemistry and biological effects of dietary phenolic compounds: relevance to cardiovascular disease. Clin Exp Pharmacol Physiol 27:152–159
Zhao Z, Egashira Y, Sanada H (2004) Ferulic acid is quickly absorbed from rat stomach as the free form and then conjugated mainly in liver. J Nutr 134:3083–3088
Surh YJ (2003) Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 3:768–780
Ippoushi K, Takeuchi A, Azuma K (2009) Prevention of peroxynitrite-induced oxidation and nitration reactions by ellagic acid. Food Chem 112:185–188
Hannum SM (2004) Potential impact of strawberries on human health: a review of the science. Crit Rev Food Sci Nutr 44:1–17
Heur YH, Zeng W, Stoner GD, Nemeth GA, Hilton B (1992) Synthesis of ellagic acid O-alkyl derivatives and isolation of ellagic acid as a tetrahexanoyl derivative from Fragaria ananassa. J Nat Prod 55:1402–1407
Mandal S, Ahuja A, Shivapurkar NM, Cheng SJ, Groopman JD, Stoner GD (1987) Inhibition of aflatoxin B1 mutagenesis in Salmonella typhimurium and DNA damage in cultured rat and human tracheobronchial tissues by ellagic acid. Carcinogenesis 8:1651–1656
Papoutsi Z, Kassi E, Tsiapara A, Fokialakis N, Chrousos GP, Moutsatsou P (2005) Evaluation of estrogenic/antiestrogenic activity of ellagic acid via the estrogen receptor subtypes ERα and ERβ. J Agric Food Chem 53:7715–7720
Yuce A, Ateşşahin A, Ceribası AO, Aksakal M (2007) Ellagic acid prevents cisplatin induced oxidative stress in liver and heart tissue of rats. Basic Clin Pharmacol Toxicol 101:345–349
Turk G, Atessahin A, Sonmez M, Ceribas AO, Yuce A (2008) Improvement of cisplatin induced injuries to sperm quality, the oxidant–antioxidant system, and the histologic structure of the rat testis by ellagic acid. Fertil Steril 89:1474–1481
Yu Y-M, Chang W-C, Wu C-H, Chiang S-Y (2005) Reduction of oxidative stress and apoptosis in hyperlipidemic rabbits by ellagic acid. J Nutr Biochem 16:675–681
Atessahin A, Ceribaş AO, Yuce A, Bulmus O, Ckim G (2007) Role of ellagic acid against cisplatin-induced nephrotoxicity and oxidative stress in rats. Basic Clin Pharmacol Toxicol 100:121–126
Hwang JM, Cho JS, Kim TH, Lee YI (2010) Ellagic acid protects hepatocytes from damage by inhibiting mitochondrial production of reactive oxygen species. Biomed Pharmacother 64:264–270
Rogerio AP, Fontanari C, Borducchi E, Keller AC, Rusco M, Soares EG, Albuquerque DA, Faccioli LH (2008) Anti-inflammatory effects of Lafoensia pacari and ellagic acid in a murine model of asthma. Eur J Pharmacol 580:262–270
de Boer JG, Yang H, Holcroft J, Skov K (2004) Chemoprotection against N-nitrosomethylbenzylamine-induced mutation in the rat esophagus. Nutr Cancer 50:168–173
Ahn D, Putt D, Kresty L, Stoner GD, Fromm D, Hollenberg PF (1996) The effects of dietary ellagic acid on rat hepatic and esophageal mucosal cytochromes P450 and phase II enzymes. Carcinogenesis 17:821–828
Shepherd AG, Manson MM, Ball HW, McLellan LI (2000) Regulation of rat glutamate-cysteine ligase (γ-glutamylcysteine synthetase) subunits by chemopreventive agents and in aflatoxin B1-induced preneoplasia. Carcinogenesis 21:1827–1834
van der Logt EM, Roelofs HM, Nagengast FM, Peters WH (2003) Induction of rat hepatic and intestinal UDP-glucuronosyltransferases by naturally occurring dietary anticarcinogens. Carcinogenesis 24:1651–1656
Baumann F, Preiss R (1973) Cyclophosphamide and related anticancer drugs. J Chromatogr B 764:173–192
Fleming RE (1997) An overview of cyclophosphamide and ifosfamide pharmacology. Pharmacotherapy 17:1465–1545
Perini P, Calabrese M, Rinaldi L, Gallo P (2007) The safety profile of cyclophosphamide in multiple sclerosis therapy. Expert Opin Drug Saf 6:183–190
Uber WE, Self SE, Van Bakel AB, Pereira NL (2007) Acute antibody-mediated rejection following heart transplantation. Am J Transplant 7:2064–2074
IARC (1987) IARC monograph on the evaluation of carcinogenicity: an update of IARC monographs 1 to 42. International Agency for Research on Cancer Supplement 7
Hales BE (1982) Comparison of the mutagenicity and teratogenicity of cyclophosphamide and its active metabolites, 4-hydroxycyclophosphamide, phosphoramide mustard and acrolein. Cancer Res 42:3016–3021
Patel JM (1987) Stimulation of cyclophosphamide-induced pulmonary microsomal lipid peroxidation by oxygen. Toxicology 45:79–91
Tripathi DN, Jena GB (2008) Astaxanthin inhibits cytotoxic and genotoxic effects of cyclophosphamide in mice germ cells. Toxicology 27:96–103
Travis LB, Curtis RE, Glimelius B, Holowaty EJ, Van Leeuwen FE, Lynch CF, Hagenbeek A, Stovall M, Banks PM, Adami J et al (1995) Bladder and kidney cancer following cyclophosphamide therapy for non-Hodgkin’s lymphoma. J Natl Cancer Inst 87:524–530
Sladek N (1971) Metabolism of cyclophosphamide by rat hepatic microsomes. Cancer Res 1:901–908
Sladek NE (1988) Metabolism of oxazaphosphorines. Pharmacol Ther 37:301–355
Roy P, Yu LJ, Crespi CL, Waxman DJ (1999) Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P-450 profiles. Drug Metab Dispos 27:655–666
Bagley C, Bostick F (2001) Clinical pharmacology of cyclophosphamide. Cancer Res 33:226–235
Lin SS, Hung CF, Ho CC, Liu YH, Ho HC, Chung JG (2000) Effects of Ellagic acid by oral administration on N-acetylation and metabolism of 2-aminofluorene in rat brain tissues. Neurochem Res 25(11):1503–1508
Suzuki N, Masamune A, Kikuta K, Watanabe T, Satoh K, Shimosegawa T (2009) Ellagic acid inhibits pancreatic fibrosis in male Wistar Bonn/Kobori rats. Dig Dis Sci 54(4):802–810
Levy L, Harris R (1977) Effect of N-acetyl cysteine on some aspects of cyclophosphamide induced toxicity and immunesupression. Biochem Pharmacol 26(11):1015–1020
Chakraborty P, Sk UH, Murmu N, Das JK, Pal S, Bhattacharya S (2009) Modulation of cyclophosphamide-induced cellular toxicity by diphenylmethyl selenocyanate in vivo, an enzymatic study. J Cancer Mol 4(6):183–189
Nafees S, Tanveer S, Wani A, Rashid S, Ali N and Sultana S (2011) Modulatory effects of gentisic acid against genotoxicity and hepatotoxicity induced by cyclophosphamide in Swiss albino mice. J Pharm Pharmacol 64(2):259–267
Schmid W (1975) The micronucleus test. Mutat Res 31:9–15
Gollapudi B, McFadden LG (1995) Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test. Mutat Res 347:97–99
Tahir M, Sultana S (2011) Chrysin modulates ethanol metabolism in Wistar rats: a promising role against organ toxicities. Alcohol Alcohol 46:383–392
Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR (1974) Bromobenzene induced liver necrosis: protective role of glutathione and evidence for 3,4-bromobezene oxide as the hepatotoxic metabolite. Pharmacology 11:151
Carlberg I, Mannervik B (1985) Glutathione level in rat brain. J Biol Chem 250:4480–4575
Mohandas M, Marshall JJ, Duggin GG, Horvath JS, Tiller D (1984) Differential distribution of glutathione and glutathione related enzymes in rabbit kidney. Cancer Res 44:5086–5091
Habig WH, Pabst MJ, Jakoby WB (1974) Glutathione-S-transferases: the first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139
Wright JR, Colby HD, Miles PR (1981) Cytosolic factors which affect microsomal lipid peroxidation in lung and liver. Arch Biochem Biophys 206:296–304
Stripe F, Della Corte E (1969) The regulation of rat liver xanthine oxidase. J Biol Chem 244:3855–3863
Claiborne A (1985) Catalase activity. In: Greenwald RA (ed) CRC handbook of methods in oxygen radical research. CRC Press, Boca Raton, pp 283–284
Kornberg A (1955) Lactic dehydrogenase of muscle. In: Colowick SP, Kaplan NO (eds) Methods in enzymology, vol I. Academic Press, New York, pp 441–443
Lowry OH, Rosebrough NJ, Farr A, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Hengstler JG, Hengst A, Fuchs J, Tanner B, Pohl J, Oesch F (1997) Induction of DNA cross links and DNA strand lesions by cyclophosphamide after inactivation by cytochrome P-450 2B1. Mutat Res 373:215–223
Haque R, Bin-Hafeez B, Parvez S, Pandey S, Sayeed I, Ali M, Raisuddin S (2003) Aqueous extract of walnut (Juglans regia L.) protects mice against cyclophosphamide-induced biochemical toxicity. Hum Exp Toxicol 22:473–480
Selvakumar E, Prahalathan C, Mythili Y, Varalakshmi P (2005) Mitigation of oxidative stress in cyclophosphamide-challenged hepatic tissue by dl-alpha-lipoic acid. Mol Cell Biochem 272:179–185
Cooper JA, Merrill WW, Reynolds HY (1986) Cyclophosphamide modulation of bronchoalveolar cellular populations and macrophage oxidative metabolism: possible mechanisms of pulmonary pharmacotoxicity. Am Rev Respir Dis 134:108–114
Kehrer JP, Biswal SS (2000) The molecular effects of acrolein. Toxicol Sci 57:6–15
Tripathi DN, Jena GB (2009) Intervention of astaxanthin against cyclophosphamide-induced oxidative stress and DNA damage: a study in mice. Chem Biol Interact 180:398–406
Bhatia K, Kaur M, Atif F, Ali M, Rehman H, Rahman S, Raisuddin S (2006) Aqueous extract of Trigonella foenum-graecum L. ameliorates additive urotoxicity of buthionine sulfoximine and cyclophosphamide in mice. Food Chem Toxicol 44:1744–1750
Cagler K, Kinalp C, Arpaci F, Turan M, Saglam K, Ozturk B, Komurcu S, Yavuz, Yenicesu M, Ozet A, Vural A (2002) Cumulative prior dose of cisplatin as a cause of the nephrotoxicity of high-dose chemotherapy followed by autologous stem-cell transplantation. Nephrol Dial Transplant 17:1931–1935
Abrahama P, Indiranib K, Sugumar E (2007) Effect of cyclophosphamide treatment on selected lysosomal enzymes in the kidney of rats. Exp Toxicol Pathol 59:143–149
Salamone MF, Heddle JA (1983) The bone marrow micronucleus assay: rationale for a revised protocol. In: de Serres FJ (ed) Chemical mutagens: principles and methods for their detection, vol 8. Plenum, New York, pp 111–149
Al-Majed AA, Al-Yahya AA, Al-Bekairi AM, Al-Shabanah OA, Qureshi S (2006) Studies on the cytological and biochemical effects of valerian in somatic and germ cells of Swiss albino mice. Food Chem Toxicol 44:1830–1837
Acknowledgment
The authors are thankful to CCRUM (AYUSH), Ministry of Health and Family Welfare, Govt. of India for providing funds to carry out this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rehman, M.U., Tahir, M., Ali, F. et al. Cyclophosphamide-induced nephrotoxicity, genotoxicity, and damage in kidney genomic DNA of Swiss albino mice: the protective effect of Ellagic acid. Mol Cell Biochem 365, 119–127 (2012). https://doi.org/10.1007/s11010-012-1250-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1250-x