
Abstract
MicroRNAs (miRNAs) are increasingly reported to have important roles in diverse biological and pathological processes. Changes in abundance of muscle-specific microRNA, miR-1, have been implicated in cardiac disease, including arrhythmia and heart failure. However, the specific molecular targets and cellular mechanisms involved in the miR-1 function in the heart are only beginning to emerge. In this study, we investigated miR-1 expression and its potential role in the mouse model of viral myocarditis (VMC). The expression levels of miR-1 and its target gene Connexin 43 (Cx43) were measured by real-time PCR and western blotting, respectively. The miR-1 expression levels were significantly increased in cardiac myocytes from VMC mice in comparison with control samples (relative expression: 10 ± 2.5 vs. 31 ± 7.6, P < 0.05). Among the target genes of miR-1, the expression Cx43 protein was significantly reduced in such mice while there was no significant difference in the its mRNA levels. Our results revealed an inverse correlation between miR-1 levels and Cx43 protein expression in VMC samples. Using a bioinformatics-based approach, we found two identical potential binding sites were found in mouse miR-1 and Cx43 3′- untranslated region, this confirms a possible regulatory role of miR-1. In cultured, miRNA transfected myocardial cells, we show overexpression of miR-1 accompanied by a decrease in Cx43 protein’s expression. There was only a slight (not statistically significant) drop in Cx43 mRNA levels. Our results indicate that miR-1 is involved in VMC via post-transcriptional repression of Cx43, and might constitute potentially valuable data for the development of a new approach in the treatment of this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Viral myocarditis (VMC), a disease without reliable or effective treatment, causes chronic dilated cardiomyopathy or death in up to 20% of affected children and 50% of affected adults [1]. The events producing cytopathic effects and leading to cardiac dysfunction after viral attachment and replication are not clearly understood. Multiple mechanisms have been implicated (reviewed previously [2]), including direct viral injury and persistence, autoimmune phenomena, cytokine fluxes, and T-cell mediated inflammatory responses. Several studies revealed that microRNAs (miRNAs) are essential for cardiac development and function. Moreover, genetic studies have identified distinct roles for specific miRNAs during cardiogenesis, cardiac hypertrophy, and electrical conduction. This led us to believe that miRNAs might also play a critical role in the pathogenesis of VMC.
MiRNAs are endogenous, 20–23 nucleotide (nt), small noncoding RNAs that negatively regulate gene expression in various eukaryotic organisms. They are transcribed as long primary transcripts (pri-miRNAs) and trimmed in the nucleus by the RNase III-type protein (Drosha/DGCR8) into approximately 70 nt stem-loop forms known as precursor miRNAs (pre-miRNAs). These are exported by exportin-5 into the cytoplasm, where they are cut by another RNase III-type protein (Dicer/TRBP) into 20–23 nt mature miRNAs. Mature miRNAs are incorporated into the RNA-induced silencing complex (RISC) which inhibits gene expression by cleaving mRNAs or blocking their translation [3–5].
MiRNAs have been reported to play important roles in diverse biological and pathological processes, including cell differentiation, proliferation, apoptosis, heart disease, neurological disorders, and human cancers [6–11]. Among the known miRNAs, miR-1 is specifically expressed in adult cardiac and skeletal muscle tissues [12–15]. Recently, miR-1 has been found to be upregulated in various cardiovascular diseases. Yang et al. [16] found that miR-1 affects Connexin 43 (Cx43) in myocardial cells in coronary atherosclerosis disease [16]. However, the expression of miR-1 in the process of VMC is not completely understood.
Gap junctions are responsible for the cell-to-cell electrical coupling required for synchronized cardiac contraction. Gap-junction channels permit the exchange of ions, small signaling molecules, and metabolites between adjacent cells [17]. Cx43 is the major gap-junction protein expressed in the heart and is responsible for the proper function of the cardiac conduction system [18].
In this study, we assess the expression of miR-1 and Cx43 in a Sprague–Dawley rat model of VMC, and investigate their potential roles in the pathology of this disease. The expression of Cx43 protein and its mRNA was also examined in miR-1 transfected, cultured myocardial cells.
Materials and methods
Mouse model of VMC
We used CVB3 viral stock derived from the infectious cDNA copy of the cardiotropic Nancy strain, as previously described [19]. All experimental procedures were in compliance with the NIH Guide for the care and use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Fudan University. Thirty BALB/c mice (male, 4–6 weeks old, 16–20 g) were infected intraperitoneally with 5 × 104 plaque-forming units of purified CVB3, as described previously [20]. Ten mice were infected intraperitoneally with MEM Eagle as control. After 15 days, the mice were killed by an intraperitoneal injection of pentobarbital (40 mg/kg) and hearts were collected for analysis. Histology was performed on transverse tissue sections covering the right and left ventricle, taken below the level of the valves, fixed in 4% phosphate-buffered paraformaldehyde (pH 7.2) and embedded in paraffin. The remainders of the left and right ventricles were snap frozen in liquid nitrogen and used for proteomic analysis.
Histological analysis
Samples were embedded in phosphate-buffered paraformaldehyde and cut into 4-μm thick sections. These were mounted on glass slides and stained with hematoxylin and eosin (H&E) for histological examination.
RNA isolation
Total RNA was extracted with TRIzol, according to the manufacturer’s guidelines (Invitrogen). Any remaining DNA was removed with the DNA-free kit (Ambion) and RNA was re-purified with an RNeasy kit, following the manufacturer’s protocol (Qiagen).
Detection of microRNA-1 and Cx43 mRNA by real-time PCR assay
For quantification of Cx43 transcript, conventional real-time RT–PCR was carried out. The total RNA samples were extracted from left ventricle or from cultured myocardial cells, as described below. TaqMan quantitative assay was performed with GAPDH expression as an internal control.
The total RNA was used to detect mature miR-1 expression using Hairin-itmiRNAs real-time PCR kit (Shanghai GenePharma, China). In brief, the real-time miRNAs assay has two steps: stem-loop RT reaction and real-time PCR detection. Stem-loop RT primers bind to the 3′ end of miRNA molecules and are transcribed with reverse transcriptase. RT product is quantified using real-time PCR, using miRNA-specific forward primer, reverse primer, and a carboxyfluorescein dye-labeled reporter probe. To normalize RNA content, the U6 snRNA was used as the internal control. The relative miR-1 expression levels in each group were calculated by the mathematical delta–delta method [21]. Reactions were run three times for each group. Specific primers were as follows:
miR-1 forward: 5′-TCAATCTCTAACAAGCTAATCTCT-3′,
miR-1 reverse: 5′-TTGACAGTAGGTTAATCCAAAGT-3′ (296 bp);
U6 forward: 5′-ATGACGTCTGCCTTGGAGAAC-3′,
U6 reverse: 5′-TCAGTGTGCTACGGAGTTCAG-3′ (291 bp);
GAPDH forward: 5′-CTGACATGCCGCCTGGAGA-3′,
GAPDH reverse: 5′-ATGTAGGCCATGAGGTCCAC-3′ (260 bp);
Cx43 NM_000165 forward: 5′-CAGCGTGACTTCACTACT-3′,
Reverse: 5′-CAGCATTGACACCATCAG-3′.
The predicted target genes and their miRNA binding site seed regions were investigated using TargetScan (release 5.1, http://www.targetscan.org/). The sequences of the predicted mature miRNAs were confirmed using miRBase (release 16.0, September 2010; http://microrna.sanger.ac.uk/).
Immunohistochemical analysis of Cx43 protein
After standard deparaffination and rehydration, specimens were exposed to xylol for 10 min, 100% alcohol for 5 min, 96% alcohol for 5 min, and 70% alcohol for 3 min. Endogenous peroxidase activity was quenched by exposure to 3% hydrogen peroxide for 10 min. The antigen was restored by immersion in citrate-buffered (pH 6.0). Normal goat serum was added for 10 min at room temperature, and Cx43 antibody (Millipore, USA) at 37°C for 1.5 h, followed by a wash with phosphate-buffered saline (PBS). Biotinylated goat anti-rabbit IgG was added for 20 min at 37°C, followed by a PBS wash. The specimens were incubated with streptavidin–biotin–peroxidase complex for 20 min at 37°C and then rinsed with PBS. The color was developed with diaminobenzidine at room temperature, then the samples rinsed with distilled water—the reactive time was controlled under the light microscope. The sections were re-stained with hematoxylin, incubated at 37°C, and sealed with neutral gum. In the negative control, the primary monoclonal antibody was omitted and replaced with PBS. Slides were examined under a light microscope.
Western blotting for Cx43 expression
The protein extract (15 μg) prepared from tissue samples or cultured heart cells was separated by SDS–PAGE and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% milk in Tris-buffer saline solution (pH 7.6) containing 0.05% Tween-20 (TBS/T), incubated with specific antibodies to Cx43 (Millipore, USA) overnight at 4°C, and then with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. The immunoreactive proteins were visualized using ECL plus detection reagents (GE Healthcare, UK). Western blot bands were quantified using Odyssey v1.2 software, by measuring the band intensity (area × OD) for each group and normalizing to GAPDH (anti-GAPDH antibody from Kangcheng Inc.) as an internal control [22].
Cell isolation from neonatal rat heart and primary cell culture
Neonatal rat ventricular cardiomyocytes were isolated and cultured as described previously [23]. In brief, 1–3 days old rats were decapitated and their hearts were aseptically removed. The ventricles were dissected, minced, and trypsinized overnight at 4°C. On the next day, the cells were dissociated with collagenase and pre-plated twice for 60 min, at 37°C. The non-adherent cardiomyocytes were removed and plated in 24-well plates in DMEM/F-12 medium (Invitrogen) containing 10% FBS and 0.1 mM bromodeoxyuridine (Sigma). 1 × 105 cells/well were seeded in six well plates for further experiments. This procedure yielded cultures with 90 ± 95% myocytes, as assessed by microscopic observation of cell beating.
Synthesis of miRNAs and sequences of miRNA inhibitor
Mouse miR-1 and its mutant RNA oligos were synthesized by Integrated DNA Technologies, Inc. The sequence of anti-miR-1 antisense inhibitor oligonucleotide-1 (AMO-1) is the exact antisense of the mature miRNA sequence (5′-ATACACACTTCTTTTACATTCCA-3′). The sequence of mis-AMO-1 (negative control) carries ten mismatched nucleotides to miR-1 primarily at the 3′-end (5′-CGCTACACTTCTTTTATCGGTTA-3′: the mismatched nucleotides are underlined). AMO-1 and mis-AMO-1 contain 2′-O-methyl modifications at every base and a 3′ C3-containing amino linker.
Transfection of miRNAs
MiR-1 and its mutant constructs were synthesized by Integrated DNA Technologies, Inc. The sequences of anti-miR-1 antisense inhibitor oligonucleotides (AMOs) used in our studies are the exact antisense copies of mature miRNA sequences, have 2′-O-methyl modifications at every base and a 3′C3 containing amino linker [16, 24]. Oligo transfection was performed according to an established protocol [25]. The cells were transfected using transfection reagent (Qiagen, Valencia, CA) for 6 h. Transfection complexes were prepared according to the manufacturer’s instructions, and 2′OMe-miR-1 or control oligo 2′OMe-EGFP was added to a final oligonucleotide concentration of 30, 50, and 100 nmol/l. The transfection efficiency was low at 30 or 50 nmol/l. However, at 100 nmol/l concentration, transfection efficiency is at least 80%; this concentration was chosen for our tests. The transfection medium was replaced with the normal culture medium after 6 h incubation.
Statistical analysis
Statistical analysis was performed using SPSS 10.5. Data are presented as mean ± standard deviation (SD). Differences between groups were assessed using one-way analysis of variance (ANOVA); P < 0.05 indicates significance. All experiments were carried out at least three times.
Results
Establishing an animal model of VMC (evidence of VMC)
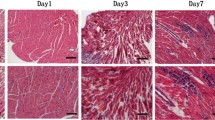
Signs of VMC were apparent in the experimental groups 3 days after virus inoculation; these included coat ruffling, weakness, and irritability. On day 3, a few scattered small foci of myocyte necrosis were noted. Myocardial necrosis and cell infiltration were extensive on day 7, with necrotic areas appearing more prominent. There were many lymphocytes and macrophages in and around the necrotic foci. Infiltration of the inflammatory cells and necrotic areas decreased, and necrotic myocardium gradually changed to fibrosis and calcification on day 15 (Fig. 1a, b). There were no necrotic lesions or signs of cell infiltration in the hearts of uninfected control mice.

a H&E stain of the paraffin-embedded left ventricle that control group (original magnification ×100). b H&E stain of the paraffin-embedded left ventricle. Many lymphocytes and macrophages infiltrate in the myocardial cells on day 3 (original magnification ×100). c H&E stain of the paraffin-embedded left ventricle. Infiltration of the inflammatory cells and necrotic areas were decreased, and necrotic myocardium gradually changed to fibrosis on day 15 (original magnification ×200)
Expression of Cx43 protein in VMC mice
We performed immunohistochemistry and western blotting of Cx43 protein. In control hearts, Cx43 immunostaining was mostly localized at the intercalated discs, (indicated by the arrows in the figure), whereas in VMC hearts Cx43 expression was diminished and relocated to lateral surface membrane (Fig. 2a). We also found that the expression of Cx43 protein decreases significantly 15 days after virus inoculation (P < 0.05, Fig. 2b).

a Immunohistochemical verification of repression of Cx43 by miR-1. In control heart, Cx43 immunostaining was mostly localized at the intercalated discs as indicated by the arrows whereas in VMC, Cx43 expression was diminished and relocated to lateral surface membrane (original magnification ×400) b Comparisons of Cx43 protein levels between membrane samples isolated from mice with healthy control hearts and from mice with VMC, determined by western blot analysis. Shown are mean ± SE from 10 Ctl and 30 VMC; *P < 0.05 versus Ctl.; unpaired student t test. c There was slightly decreased expression of Cx43 at the mRNA levels (*P > 0.05 vs. Ctl; unpaired student’s t test.). Results were normalized to expression of GAPDH
Notably, the changes of Cx43 expression at the mRNA level did not reflect the protein expression level. Quantitative RT–PCR demonstrated much smaller decrease in the level of Cx43 transcript than that of its protein (Fig. 2c). The Cx43 protein expression diminished nearly threefold (P < 0.05) but only 30% (P > 0.05) drop in the mRNA levels was observed.
These results suggest a post-transcriptional mechanism mediating the expression regulation of Cx43 gene in VMC mice.
MiR-1 expression in the mouse model of VMC
The miRNAs’ mechanism of action involves incorporation of the single-stranded miRNA into the RISC and its subsequent binding to the 3′-UTR of the target mRNA through exact complementarity with the 5′ end’s 7–8 nt, and partial complementarity with the rest of the sequence [26–29]. In this way, miRNAs achieve translational inhibition. To explore the possible involvement of miRNAs in the regulation of Cx43 expression, we used a bioinformatics-based approach to predict the putative targets, employing TargetScan hosted by the Wellcome Trust Sanger Institute [30]. Two identical potential binding sites were found for mouse miR-1 and Cx43 (Fig. 3a). We found that the expression level of miRNA-1 increased by approximately 200% in cardiac ventricle tissues of VMC mice (relative expression: 10 ± 2.5 vs. 31 ± 7.6, P < 0.05, Fig. 3b).

a The complementary sequences between miR-1 and its putative sites within the 3′-UTRs of mouse Cx43 mRNAs, predicted with computational and bioinformatics-based approach using TargetScan hosted by Wellcome Trust Sanger Institute. Watson–Crick complementarity was shown in bold and connected by “|”. Note that there are two putative miR-1 target sites in the 3′-UTR of Cx43. b miR-1 expression was greatly increased in VMC mice (*P < 0.05). Results were normalized to expression of U6
Post-transcriptional repression of Cx43 by miR-1
The above results show that both the expression of Cx43 protein and of miR-1 change in VMC mice. We also evaluated the ability of miR-1 to repress Cx43 expression in cardiac myocytes. As depicted in Fig. 4a, transfection of miR-1 into the cultured cardiac myocytes can substantially reduce Cx43 protein levels. Transfection with mis-miR-1 (negative control) did not affect Cx43 expression. Moreover, when we co-transfected the anti-miR-1 antisense inhibitor oligonucleotide (AMO-1) with miR-1, it nearly abolished the effect of miR-1, thus, verifying the specificity of the miR-1 action. Co-transfection of mis-AMO-1 (negative control) with miR-1 reduced the levels of Cx43 protein (Fig. 4c), i.e., no cancellation of miR-1 effect was observed. By comparison, miR-1 produced virtually no effect on Cx43 mRNA levels (Fig. 4d), indicating that it does not affect Cx43 mRNA stability.

Post-transcriptional repression of Cx43 by miR-1. a MiR-control (negative control), miR-1, and AMO-1 were transduced into neonatal rat cardiac myocytes. The transduction efficiency, which is shown in the picture, was always over 80%. b Levels of miR-1 in cultured cardiac myocytes that were transfected with miR-1 alone or with miR-1 and mis-miR-1, AMO-1, or mis-AMO-1. Data are mean ± SE (n = 5 independent samples for each group); *P < 0.05 versus Ctl, # P > 0.05 versus Ctl; unpaired student’s t test. c Cx43 protein levels were determined by western blotting of membrane samples from cultured neonatal rat ventricular myocytes [31]. Cells were transfected with miR-1 alone or with miR-1 and mis-miR-1, AMO-1 or mis-AMO-1. AMO-1 reversed the repressing effects of miR-1 on Cx43, but mis-AMO-1 did not. Mean ± s.e.m. from five batches of cells. *P < 0.05 versus Ctl, + P < 0.05 versus miR-1 alone; unpaired student’s t test. d Quantitative real-time PCR analysis showed no significant change in mRNA levels of Cx43 in miR-1 over-expressing cardiac myocytes. Values are the means ± S.E. of 5–6 independent experiments (n.s. not significant)
Discussion
Viral myocarditis remains a significant clinical challenge because the pathogenesis of the disease is poorly understood, morbidity and mortality are high, and currently employed treatment strategies have little impact on improving the outcome. Ongoing myocarditis is often caused by infection with Coxsackie viruses of group B (CVB), the members of the enterovirus genus of the Picornaviridae [32]. Even though it is accepted that CVB3 is a causative agent of myocarditis, the actual mechanisms controlling the pathogenesis and outcome of the disease have not been resolved yet. Currently, three different mechanisms of VMC pathogenesis are suggested: (i) injury of cardiomyocytes directly induced by CVB3, (ii) damage to the myocardium triggered by excessive reaction of infiltrating immune cells, and (iii) impairment of myocardial function caused by cardiac auto-antibodies, possibly as a result of molecular mimicry [33]. The question arises whether miRNA also plays a critical role in the pathogenesis of VMC. Taking into account our own recent results and the data found in existing publications, we suggest a possible role for microRNAs during the VMC initiation and progression, involving an inappropriate repression or de-repression of crucial protein targets.
Changes in miR-1 levels have been reported for several heart diseases but the data vary substantially among the studies published: both increases and decreases in miR-1 expression were demonstrated. Sayed et al. [34] shows that miR-1 is downregulated in two different murine models of cardiac hypertrophy and cardiac failure. MiR-1 expression also decreases in dilated cardiomyopathy, atrial fibrillation and aortic stenosis [35, 36]. However, there are some reports of increased miR-1 expression in human hearts from patients with diabetes mellitus [37]. Yang et al. [16] demonstrates upregulation of the miR-1 in hearts from myocardial infarction patients and provide evidence that miR-1 mediates electrical remodeling and arrhythmias. The available data present a complex picture of possible regulatory mechanisms and strongly suggest miR-1 as a good candidate both for a diagnostic biomarker and a therapeutic target in many heart diseases.
The results of our study show Cx43 immunostaining mostly localized at the intercalated discs of healthy heart, whereas in VMC the expression of Cx43 is diminished and relocated to lateral surface membrane. Using western blotting, we found that Cx43 expression was greatly decreased in VMC mice. We also show that the reduction of Cx43 expression is associated with increased levels of inhibitor miR-1. Given our results, we can assume that overexpression of miR-1 causes post-transcriptional repression of Cx43 synthesis in cardiac myocytes. However, we observe only a slight decrease in Cx43 mRNA levels both in murine VMC models and in miR-1 overexpressing, cultured cardiac myocytes.
Yang et al. [16] inserted the 3′-UTR of GJA1, encoding gap-junction protein Cx43, into the 3′-UTR of a luciferase reporter plasmid containing a constitutively active promoter, and verified that the 3′-UTR of GJA1 contain nucleotides that are complementary to the 5′ end of miR-1. Our results are in concordance with theirs; we also demonstrate that miR-1 can post-transcriptionally repress the expression of the gap-junction protein Cx43 in cardiac myocytes. Our study confirms this miRNA’s involvement in VMC; we believe that the increased levels of miR-1 are likely to be implicated in the pathogenesis of VMC.
Connexin 43 is the main protein forming gap-junction channels in ventricular myocardium, allowing electrical coupling and communication between adjacent cardiomyocytes [38, 39]. It plays an important role in the pathophysiology of VMC. Altered expression or distribution of Cx43 might impair propagation of the electrical impulse and favor the appearance of arrhythmias,sometimes even leading to sudden death [38, 39].
On the basis of our results, it appears reasonable to propose that the expression of miR-1 is upregulated in VMC disease via some unknown mechanisms and the increased miR-1 levels can cause a downregulation of Cx43 protein’s synthesis. In our experiments, miR-1 did not significantly affect the mRNA levels of Cx43, possibly due to very imperfect complementarities between the miRNAs and the genes. Respecting Cx43 protein is an important transmembrane protein in cardiac myocytes, the decreased Cx43 may lead to interferencing cardiac function.
Our study reveals the ability of a miRNA to regulate the expression of an ion channel protein in VMC. Thus, it expands our understanding of the cellular function and pathophysiological roles of miRNAs in general, confirming the view that miRNAs are likely to have many widespread and varied functions in the cell. Further research into the function of specific miRNAs in heart development and disease, and identification of their target molecules, should help us to understand the molecular mechanisms of those processes. Hopefully, increasing the knowledge in this field will provide new target genes for the treatment of cardiac diseases.
References
Feldman AM, McNamara D (2000) Myocarditis. N Engl Med 343:1388–1398
Miyamoto SD, DeBiasis RL, Long CS (2008) Novel therapeutic targets in viral myocarditis. Future Virol 3:373–381
Tsuchiya S, Okuno Y, Tsujimoto G (2006) MicroRNA: biogenetic and functional mechanisms and involvements in cell differentiation and cancer. J Pharmacol Sci 101(4):267–270
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Karp X, Ambros V (2005) Developmental biology: encountering microRNAs in cell fate signaling. Science 310:1288–1289
Kloosterman WP, Plasterk RH (2006) The diverse functions of microRNAs in animal development and disease. Dev Cell 11:441–450
Wang Y, Stricker HM, Gou D, Liu L (2007) MicroRNA: past and present. Front Biosci 12:2316–2329
van Rooij E, Sutherland LB, Liu N et al (2006) A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103:18255–18260
Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A (2007) A microRNA feedback circuit in midbrain dopamine neurons. Science 317:1220–1224
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6:857–866
Zhao Y, Samal E, Srivastava D (2005) Serum response factor regulates a muscle specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38:228–233
Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF (2006) Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci USA 103:8721–8726
Kwon C, Han Z, Olson EN, Srivastava D (2005) MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA 102:18986–18991
Yang B, Lin H, Xiao J et al (2007) The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13:486–491
Harris AL (2007) Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol 94:120–143
Miquerol L, Dupays L, Theveniau-Ruissy M et al (2003) Gap junctional connexins in the developing mouse cardiac conduction system. Novartis Found Symp 250:80–98 Discussion 98–109:276–279
Klingel K, Hohenadl C, Canu A, Albrecht M et al (1992) Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA 89:314–318
Kandolf R, Selinka H, Klingel K et al (2002) Pathogenesis of Coxsackievirus B infections. Mol Biol of Picornaviruses 67:405–413
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Luo CL, Li BX, Xu HF et al (2011) Autophagy is involved in traumatic brain injury-induced cell death and partially contributes to functional outcome deficits in mice. Neuroscience 184:54–63
Pang L, Koren G, Wang Z, Nattel S (2003) Tissue-specific expression of two human Cav1.2 isoforms under the control of distinct 5′-flanking regulatory elements. FEBS Lett. 546:349–354
Luo X, Xiao J, Lin H et al (2007) Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol 212:358–367
Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA (2001) Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 98:9742–9747
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3:e85
Cannell IG, Kong YW, Bushell M (2008) How do microRNAs regulate gene expression? Biochem Soc Trans 36:1224–1231
Nilsen TW (2007) Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet 23:243–249
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109–D111
Salameh A, Schneider P, Mühlberg K et al (2004) Chronic regulation of the expression of gap junction proteins connexin40, connexin43, and connexin45 in neonatal rat cardiomyocytes. Eur J Pharmacol 503:9–16
Kandolf R, Selinka H, Klingel K (2002) Pathogenesis of Coxsackievirus B infections. Molecular Biology of Picornaviruses. American Society of Microbiology, Washington, DC, pp 405–413
Esfandiarei M, McManus BM (2008) Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol 3:127–155
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M (2007) MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100:416–424
Ikeda S, Kong SW, Lu J et al (2007) Altered microRNA expression in human heart disease. Physiol Genomics 31:367–373
Girmatsion Z, Biliczki P, Bonauer A et al (2009) Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 12:1802–1809
Xiao J, Luo X, Lin H et al (2007) MicroRNA miR-133 represses HERGK+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem 17:12363–12367
Desplantez T, Dupont E, Severs NJ et al (2007) Gap junction channels and cardiac impulse propagation. J Membr Biol 218:13–28
Jansen JA, van Veen TA, de Bakker JM et al (2010) Cardiac connexins and impulse propagation. J Mol Cell Cardiol 48:76–82
Acknowledgments
We are grateful for the support of the Key Laboratory of Viral Heart Disease, Ministry of Public Health, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital Fudan University. The study was supported by the National Science Foundation of China (No. 81172897).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, HF., Ding, YJ., Shen, YW. et al. MicroRNA- 1 represses Cx43 expression in viral myocarditis. Mol Cell Biochem 362, 141–148 (2012). https://doi.org/10.1007/s11010-011-1136-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-011-1136-3