
Abstract
CK2 is a highly conserved, ubiquitous, signal responsive protein serine/threonine kinase. CK2 promotes cell proliferation and suppresses apoptosis, and increased CK2 expression is observed in all cancers examined. We previously reported that direct injection of antisense (AS) CK2α phosphorothioate oligonucleotides (PTO) into xenograft prostate tumors in mice significantly reduced tumor size. Downregulation of CK2α in tumor cells in vivo appeared to result in overexpression of CK2α′ protein. This suggested that in cancer cells downregulation of CK2α might be compensated by CK2α′ in vivo, prompting us to design a bispecific (bs) AS PTO (bs-AS-CK2) targeting both catalytic subunits. bs-AS-CK2 reduced CK2α and α′ protein expression, decreased cell proliferation, and induced apoptosis in cultured cells. Biodistribution studies of administered bs-AS-CK2 oligonucleotide demonstrated its presence in orthotopic prostate xenograft tumors. High dose injections of bs-AS-CK2 resulted in no damage to normal liver or prostate, but induced extensive cell death in tumor tissue. Intraperitoneal treatment with bs-AS-CK2 PTO decreased orthotopic tumor size and downregulated both CK2 mRNA and protein expression. Tumor reduction was accomplished using remarkably low doses and was improved by dividing the dose using a multi-day schedule. Decreased expression of the key signaling pathway proteins NF-κB p65 and AKT was also observed. We propose that the molecular downregulation of CK2 through bispecific targeting of the two catalytic subunits may be uniquely useful for therapeutic elimination of tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Work spanning more than three decades has culminated in establishing the profound importance of protein kinase CK2 in cell functions, including those in cancer pathobiology [1–5]. The strong impact of CK2 in cells relates to its ability to phosphorylate well over 300 potential substrates involved in diverse functions, such as activities related to gene expression, cell growth, and cell death [6, 7]. CK2 is a ubiquitous serine/threonine protein kinase that is among the most highly conserved proteins in nature. Its heterotetrameric structure consists of two catalytic subunits (42 kDa α and/or 38 kDa α′) and two regulatory subunits (28 kDa β) which are present in different configurations depending on cell type. The two catalytic subunits are linked through the β subunits, which stimulate rather than inhibit the kinase activity [8]. The β subunits may form a linkage with the nuclear matrix [9]. The nuclear matrix presents a key locus for CK2 signaling in the nucleus for its functions in cell proliferation and cell death [3, 10–13].
This laboratory was the first to undertake studies of protein kinase signaling in the prostate [14, 15], which led to the identification of CK2 as an important signal in the androgen- and growth factor-mediated regulation of growth [16]. Dynamic shuttling to various loci, such as nuclear structures including chromatin and the nuclear matrix, in response to altered growth stimuli is a key feature of CK2 signaling in the cell [12, 17–24]. Given the involvement of nuclear matrix in cell cycle control, DNA repair, chromatin remodeling, and apoptosis [25, 26], it is particularly interesting that CK2 dynamics in the nuclear matrix appear to be the most remarkable indicator of the powerful influence of CK2 in cell fate decisions. For example, an early response to downregulation of CK2 expression is the rapid loss of CK2 from the nuclear matrix associated with induction of apoptosis in prostate tumor cells in culture and in vivo [27–29].
CK2 was originally shown to be remarkably elevated in neoplastic human prostate [30, 31], and subsequent work has shown it to be upregulated in all cancers examined [4, 32]. Although it has long been appreciated that CK2 is elevated during normal and cancer cell proliferation, our work established that increased amounts of CK2 in cancer cells were not simply a reflection of their proliferative status but additionally of the state of dysplasia [33]. A distinguishing feature of CK2 in cancer is that tumor cells have a high nuclear concentration of CK2, whereas in normal cells the protein is diffused in various compartments. Further, the level of dysregulation of CK2 appears to relate to the severity of disease and prognosis [34, 35], an idea reinforced by recent observations in other laboratories [36–41]. Programmed cell death or apoptosis is a key component of cell physiology, and its dysregulation is recognized as a critical factor in oncogenesis, especially in prostate cancer (PCa) progression [42]. We demonstrated that CK2 can potently suppress apoptosis mediated via diverse pathways (intrinsic and extrinsic) [1, 43]. This discovery that CK2, besides being involved in cell growth and proliferation, is a potent suppressor of apoptosis firmly ties CK2 upregulation to the cancer cell phenotype. Considering that cancer cells consistently demonstrate deregulation of both proliferative and apoptotic pathways, elevated CK2 would have a dual effect in cancer cells—promote proliferation and at the same time impede cell death. Animal studies have corroborated that even moderate aberrant expression of CK2 can impart remarkable oncogenic potential when in combination with other oncogenes [44–48]. Furthermore, recent studies demonstrated amplification and over-expression of the CK2α intronless gene (CSNK2A1P) in human lung cancer, with selective amplification of the 398T allele [49].
CK2 has been validated in mouse models and human clinical trials as an effective target for cancer therapy by numerous approaches. These approaches include the use of small molecule inhibitors to block CK2 catalytic activity and the use of a peptide that impairs CK2 substrate phosphorylation [50–54]. Further, we have shown the efficacy of simultaneously targeting both CK2 catalytic subunits in head and neck squamous cell carcinoma (HNSCC) using an anti-CK2 antisense-based nanoencapsulated therapeutic [5, 55]. PCa is initially androgen sensitive and responds to androgen ablation, but the disease relapses in a generally fatal form because of the emergence of an androgen depletion-insensitive or castration-resistant (CRPC) phenotype. At this stage, chemotherapy may improve survival, but most patients die from chemoresistant disease. Induction of apoptosis is an attractive approach that has gained much attention for the development of cancer therapeutics. We originally employed antisense (AS) CK2α oligonucleotide (OGN) to downregulate CK2 in cell culture and xenograft PCa models [27, 28]. However, subsequent analysis demonstrated increased prostatic CK2α′ expression after AS CK2α OGN treatment in vivo. This novel observation suggested the importance of simultaneously targeting both catalytic subunits of CK2 to insure that in the face of CK2α downregulation, CK2α′ does not take on a compensatory role in vivo that may potentially promote cancer cell survival.
In this body of work, we report on the use of a bispecific AS sequence which simultaneously targets both α and α′ catalytic subunits of CK2 (bs-AS-CK2) in a murine orthotopic xenograft model of PCa. The efficacy we observe using bs-AS-CK2 in these studies establishes proof-of-principle that molecular downregulation of CK2 expression is effective in treating PCa tumors.
Materials and methods
Cell lines and xenograft tumors
PC3-LN4 cells were maintained in monolayer culture containing modified Eagle’s MEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, and penicillin–streptomycin as described previously [56]. BPH-1 cells were maintained in monolayer culture containing RPMI 1640 supplemented with 10% heat-inactivated FBS and 2 mM l-glutamine [57]. Cells are grown in an incubator at 37°C with 5% CO2. All cells had undetectable levels of mycoplasma.
Oligonucleotides
For cell culture studies, phosphorothioate-modified oligodeoxynucleotides (PTOs) were obtained from Integrated DNA Technologies (Coralville, IA). For animal studies, PTO was obtained from TriLink Biotechnologies (San Diego, CA). The bispecific AS CK2 sequence (bs-AS-CK2) is 5′-ATACAACCCAAACTCCACAT-3′. The CK2α AS sequence was detailed previously [27]. Other bispecific AS sequences directed against CK2α and α′ tested were previously published [58, 59]. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) sequence was previously published [60].
Animals
Male athymic BALB/c nude mice (obtained from the Frederic Cancer Research Facility, National Cancer Institute, Frederic, MD) were maintained in a laminar airflow cabinet under pathogen-free conditions and were used at 8–12 weeks of age. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with the current regulations and standards of the U.S. Department of Agriculture, U.S. Department of Health and Human Services, and NIH. Animal experiments were conducted in the facilities of the Minneapolis VA Health Care System according to VA and University of Minnesota approved IACUC protocols.
In vivo therapy of orthotopic xenograft human prostate cancer in athymic nude mice
For the dose response and schedule response experiments, cultured PC3-LN4 cells (60–70% confluent) were prepared for injection as previously described [56]. Mice were anesthetized with isoflurane, a midline abdominal incision was made to expose the prostate, and viable tumor cells (2 × 106 per ml, 0.1 ml per mouse) were injected into the prostate. Therapy began when tumors were palpable and reached a diameter of approximately 3–5 mm (designated day 0). Groups of mice (see figure legends for numbers per group) were subjected to intraperitoneal (i.p.) injection with either a single dose or multiple doses (see figures) of bs-AS-CK2 PTO, GAPDH PTO or saline. Mice were killed after 10–13 days of therapy, and tumors were excised, weighed following removal of necrotic material, and snap frozen in liquid nitrogen for RNA extraction and protein analysis.
For the pharmacologic window experiments, mice were prepared as described for the above therapy studies. When tumors were palpable and reached a diameter of approximately 3–5 mm, mice were injected twice i.p. with 0.7 or 7.0 mg/kg bs-AS-CK2, 0.7 mg/kg AS-GAPDH, or saline with a 24 h interval between each injection. 10 days following the first injection tissues and tumor were processed for hematoxylin and eosin (H&E) staining and microscopy.
Biodistribution studies
For biodistribution studies, mice were prepared as for therapy studies and studied as described [61, 62]. Briefly, mice were injected with two doses of FITC-labeled bs-AS-CK2 (66 μg/kg) with a 24 h interval between each injection. Tissues were collected 24 h after the second dose, weighed, homogenized in lysis buffer (PBS with 0.005% Triton X-100), cleared by centrifugation (30 min, 300×g) and kept on ice until fluorometer measurement. FITC fluorescence was quantified at excitation/emission wavelength settings of 485 nm/538 nm against a standard curve. Background was subtracted using organ homogenates from mice processed equivalently but treated with PBS and dose ratios were calculated as percent of injected dose per gram of wet tissue weight.
Cell proliferation assays
For proliferation assays, PC3-LN4 cells were plated in MEM containing 0.01% heat-inactivated FBS at a density of 1500 cells per well in 96 well Primaria™ plates containing polyamine surface nanofibers (Surmodics Synthetic ECM) pre-coated overnight with human tenascin-C (Millipore CC065) and bovine fibronectin (Sigma F-1141) in a 3:1 ratio at a total concentration of 0.5 μg/cm2 in PBS (Invitrogen 14190). BPH-1 cells were plated in RPMI with 10% heat-inactivated FBS at a density of 1500 cells per well in 96 well Primaria™ plates pre-coated overnight with laminin (Sigma L-2020) at 0.5 μg/cm2 in PBS. For sequence testing, PTO was added directly to cells 24 h after plating at a concentration of 7.5 μM. At 48 h following PTO addition, the media was replaced with 100 μl fresh media containing 0.5 μCi 3H-thymidine (NEN NET027) and the cells harvested for scintillation counting after additional 24 h incubation. For all further proliferation assays using bs-AS-CK2, PTO were transfected into the cells at various concentrations using Dharmafect 2 (Dharmacon, Inc.) 24 h after plating. After 72 h, the media was replaced with 100 μl fresh media containing 0.5 μCi 3H-thymidine (NEN NET027). At 96 h, the cells are harvested onto filters and placed into scintillation vials with scintillation fluid for measurement of radioactivity.
TUNEL analysis for detection of apoptosis
PC3-LN4 cells were plated at a density of 5 × 105 cells on 60 mm plates. The following day, PTO was transfected into the cells at various concentrations using 2.5 μl of Dharmafect 2 (Dharmacon, Inc.) per 1 μg of PTO. After 5 h, the transfection mixture was replaced with fresh complete media. Cells were split as needed at 24 h and were plated onto cover slips at 48 h post-transfection. At 72 h, the cells were fixed with 3% paraformaldehyde for 30 min at 4°C, washed twice with phosphate-buffered saline, permeabilized with ice cold methanol for 10 min, and washed twice with nanopure water. TUNEL analysis was performed using the kit APO-BRDU (Phoenix Flow Systems, Inc). Cell nuclei were counterstained for 30 s in 5 μg/ml bisbenzimide, the cells washed 1× PBS (pH 7.4) with 0.05% NP-40 (PBS-NP) and 1× water for 5 min each, and mounted onto a slide in aqueous no-fade solution. The cells were viewed using an Olympus BX20 fluorescent microscope with images collected using a digital color CCD camera (SPOT Diagnostic Instruments, Ann Arbor, MI). 100 cells were analyzed per transfection condition, and the experiment was performed twice. Antibodies used: BrdU from BD Transduction Laboratories (347580); biotinylated sheep anti-mouse (515-066-072) and streptavidin-Cy3 (016-030-084) from Jackson Labs.
Immunoblot analysis of cultured cells and tumors
PC3-LN4 cultured cells were transfected as described above for TUNEL analysis. Cells were split as needed and collected at 24, 48, and 72 h post-transfection using trypsin. The cell pellets were frozen at −20°C immediately following collection. For immunoblot analysis, cell pellets were resuspended in 150 μl of RIPA lysis buffer (150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 1% Igepal CA630, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 10% glycerol) with the addition of Sigma protease (P8340) and phosphatase (P2850) inhibitor cocktails (used 1:100). The lysate was frozen at −70°C for 10 min, thawed on ice, vortexed for 5 s, and centrifuged for 5 min at 18,000×g and 4°C in a microcentrifuge. The supernatant was transferred to a fresh tube on ice, and quantitated using Bio-Rad protein assay reagent (500-0006). Equal quantities of lysates were separated in a 10% Tris–Glycine–SDS polyacrylamide gel by electrophoresis, and transferred to nitrocellulose membrane (Whatman 10 402 495) using the wet transfer method in Tris–Glycine–20% methanol. The membranes were blocked for 30 min with 5% nonfat milk (Bio-Rad 170-6404) in Tris-buffered saline (TBS, pH 7.4) with 0.1% Tween 20 (TBS-T) at room temperature. The membranes were then incubated overnight rocking at 4°C in 5% milk/TBS-T containing primary antibody. After washing 3 × 10 min in TBS-T, the membranes were incubated with secondary antibody in 5% milk/TBS-T for 1 h at room temperature and washed in TBS-T as before. Proteins were detected by enhanced chemiluminescence using Pierce SuperSignal West Pico and Dura substrates (Pierce 34078, 34076) and Blue Lite autorad film (ISC BioExpress F-9024-8×10).
Approximately 0.1 mg of tumor tissue was minced then homogenized into 1 ml of cytoskeleton (CSK) buffer and nuclear matrix was prepared as previously described using method C [18] with the following modifications. The homogenates were not filtered, and the final nuclear matrix pellets were resuspended in 250 μl RIPA lysis buffer and incubated on ice with repeated vortexing for 30 min. The suspension was centrifuged for 10 min at 18,000×g and the supernatant was transferred to a fresh 1.5 ml tube. Quantitations of cytosol and nuclear matrix were performed with the addition of H2O2 as described previously [63]. Cell fractions (20 μg of cytosol, 15 μg of nuclear matrix) were precipitated by addition of 3–5 volumes of −20°C acetone/methanol (1:1), vortex mixing, and centrifugation for 15 min at 18,000×g and 4°C. The pellet was washed with 0.5 ml −20°C methanol and centrifuged for 10 min at 18,000×g and 4°C. The protein pellet was resuspended in 20 μl of Novex loading buffer with tris(2-carboxyethyl)phosphine (TCEP) in RIPA buffer (each diluted to 1× concentration), the samples were heated at 37°C for 10 min, vortexed, and electrophoresed through NuPage 4-12% Bis–Tris 20 well gels (Invitrogen WG1402A) using MOPS SDS running buffer (Invitrogen NP0001). The gels were transferred to nitrocellulose membrane using the wet transfer method in Tris–Glycine–20% methanol. Protein detection was performed as above.
Antibodies used were Actin (sc-1616) and LDH-A (sc-27230) from Santa Cruz Biotechnology; CK2α (A300-197A) and CK2α′ (A300-199A) from Bethyl Laboratories; Casein Kinase II α/α′ from BD Transduction Laboratories (611611); CK2β (218712) from Calbiochem; Lamin B1 (33-2000) from Invitrogen; NF-κB p65 (610868) from BD Transduction Laboratories; Akt (9272) and Phospho-Akt Ser473 (9271) from Cell Signaling. Please note that in Fig. 1a, a chicken polyclonal anti-CK2αα′β antibody was used [64], however, because following this experiment this antibody was no longer functional, all subsequent immunoblots shown for CK2αα′ used the Bethyl Laboratories antibodies.

a Downregulation of CK2α in prostate tumors causes increased expression of CK2α′. Whole cell lysates from orthotopic xenograft prostate tumors treated via tail vein injection with 200 μl of 2 mg/ml AS-CK2α phosphodiester OGN in PBS or 200 μl of PBS were subjected to immunoblot analysis using antibodies directed against CK2α/α′ and actin. Signal density was quantitated using Image J and normalized to actin. Average expression in AS-CK2α treated tumors for CK2α and α′ was 0.38 ± 0.10 and 0.88 ± 0.02 compared to 0.58 and 0.24 for PBS treated tumor. b A bispecific antisense sequence reduces cell viability better than α or α′-specific sequences alone. PTO (7.5 μM) were directly administered to PC3-LN4 cells grown on tenascin and fibronectin. Cell proliferation was assessed by means of 3H-Thy incorporation over a 24 h period. Both sense-CK2α and AS-GAPDH were included as controls. bs-AS-CK2 #1 and #2 represent different bispecific OGN sequences designed to simultaneously target CK2α and α′. Data are presented as the mean ± SEM
Purification of RNA from tumors and real-time PCR
Total RNA was isolated from approximately 40 mg of tumor tissue using 1 ml Trizol reagent (Invitrogen 15596-026). The total RNA pellet was resuspended in 50 μl of RNase-free H2O and further purified using the RNeasy mini kit (Qiagen 74104) including the on-column DNase digestion step. The final RNA was quantitated using a NanoDrop machine. The Superscript III kit (Invitrogen) was used to synthesize cDNA from total RNA (1 μg) using an oligo-dT primer. The real-time reactions were run using RT2 qPCR primer assays (SABiosciences), 96 well FAST plates and an ABI 7900HT machine (Applied Biosystems Inc., Foster City, CA). Analyses were performed using ABI SDS 2.3 software. Initial experiments were performed to determine the amount of total RNA (0.5 and 1 μg input to cDNA reaction) and cDNA (100–10−3 dilution used for 1 μl input to PCR) to use in order to remain in the linear range of results. HPRT-1 was used as the reference gene for normalization. All results are reported as the average of reactions run at two different dilutions of cDNA.
Statistical analysis
The results of cell viability, proliferation, and TUNEL assays are presented as means ± SEM. The statistical analyses were performed using GraphPad Prism 5.02 (GraphPad Software). For the cell proliferation assays, statistical differences between mean values for treatment versus the control set to 1 or 100% were evaluated using the 1-tailed unpaired t test with Welch’s correction. Statistical differences between mean tumor weight values for each treatment group alone versus the control group were evaluated using one-way ANOVA with the Bonferroni comparison; the P value was not corrected for multiple comparisons. Differences were considered significant for P values less than 0.05.
Results
Increased CK2α′ expression following antisense-mediated knockdown of CK2α in vivo—selection of an α and α′ bispecific sequence
In initial xenograft experiments, we targeted CK2α using an AS oligonucleotide (OGN). In this experiment, athymic nude mice with orthotopic xenograft prostate tumors were treated via tail vein injection with AS-CK2α phosphodiester OGN. Following analysis of protein expression for both CK2α and α′ in treated tumors, we found that the CK2α showed decreased expression, whereas the CK2α′ subunit showed increased expression (Fig. 1a). We inferred this result reflected a possible compensatory mechanism employed by the PCa cells following decreased CK2α protein levels. Based on this result, we tested regions of high homology in both the α and α′ UTR and coding sequence information as AS OGNs for the ability to decrease cell proliferation following transfection into cancer cells compared to transfection of OGNs to the different CK2 subunits alone. OGN sequence information is provided in Table 1. Using 3H-Thymidine incorporation assays 72 h following addition of the OGN to carcinoma cells grown on extracellular matrix, we identified the bispecific (bs) sequence which produced the greatest reduction in cell proliferation (Fig. 1b). We refer to this sequence (shown as bs-AS-CK2 #1 in Fig. 1b) for AS chemistry as bs-AS-CK2. While the difference in proliferation effect between combined use of separate AS-CK2α and AS-CK2α′ OGNs and use of bs-AS-CK2 #1 is not great, the bispecific sequence allows the convenience and accuracy for delivery purposes of using one OGN. When bound to the complimentary mRNA sequence, this bs-AS-CK2 OGN is efficiently cleaved by RNase H in vitro (unpublished observation).
Effects of bs-AS-CK2 PTO introduction into cultured cells
We examined the efficacy of the bs-AS-CK2 PTO for knocking down CK2 catalytic subunit expression in PC3-LN4 (PC-3M-LN4) PCa cells. PC3-LN4 cells were derived from a fourth generation lymph node metastases of PC3M cells after orthotopic prostate injection in a nude mouse [56]. PC3M cells were, in turn, derived from liver metastases subsequent to intrasplenic injection of the androgen insensitive PC3 cells [65, 66]. These cells are highly tumorigenic, metastatic, and androgen insensitive [56]. Transfection of PC3-LN4 cells grown in standard cell culture conditions with 80 nM bs-AS-CK2 PTO resulted in a marked loss of CK2α protein levels to 0.16 ± 0.07 and CK2α′ protein levels to 0.75 ± 0.33 relative to the control PTO to red fluorescent protein (AS-RFP) at 72 h post-transfection (Fig. 2a). The cellular effects of bs-AS-CK2 transfection on cell proliferation were assessed in both PC3-LN4 and benign prostatic hyperplasia-1 (BPH-1) cells [57, 67]. The downregulation of CK2αα′ protein expression at 72 h post-transfection correlates with a distinct loss of cell proliferation as measured by 3H-Thymidine incorporation in cells grown on a matrix of tenascin and fibronectin for the malignant cells PC3-LN4 and laminin for the benign cells BPH-1 (Fig. 2b). A loss of cell viability of greater than 50% as measured by the WST-1 assay was also observed at 72 h post-transfection in cells grown directly on tissue culture treated plastic (data not shown). Finally, transfection of cells grown under standard cell culture conditions with bs-AS-CK2 also results in an induction of apoptosis at 72 h, as measured by TUNEL assay (Fig. 2c). Induction of apoptosis was quantitated at 34.6 ± 4.3% and 18.2 ± 2.3% following transfection of 320 and 160 nM bs-AS-CK2, respectively. The control apoptosis levels were 7.1 ± 2.0% and 0.7 ± 0.5% for 160 nM AS-GAPDH and Dharmafect reagent, respectively.

a bs-AS-CK2 downregulates both CK2α and α′ proteins in cultured PC3-LN4 prostate cancer cells. PC3-LN4 cells were transfected with 80 or 160 nM bs-AS-CK2 or 160 nM AS-RFP (red fluorescent protein) PTO using DOTAP reagent. Cells were collected at 48 and 72 h post-transfection and analyzed by immunoblot for CK2α and α′ and actin expression. b bs-AS-CK2 decreases DNA synthesis in cultured prostate cells. Cultured PC3-LN4 cells grown on tenascin and fibronectin and BPH-1 cells grown on laminin cells were transfected using Dharmafect reagent with 50 or 100 nM bs-AS-CK2 or AS-RFP PTO. After 48 h, 3H-Thy was added, and cells were harvested for counting at 72 h. *Significant difference from Dharmafect (P < 0.005). ^ Significant difference from AS-RFP (P < 0.05). c bs-AS-CK2 induces apoptosis in cultured PC3-LN4 prostate cancer cells. PC3-LN4 cells were transfected with 160 or 320 nM bs-AS-CK2 or 160 nM AS-GAPDH PTO using Dharmafect reagent. After 48 h, the cells were plated onto cover slips. Cells were fixed at 72 h post-transfection, and analyzed using a TUNEL staining kit
Localization of bs-AS-CK2 to tumors following intraperitoneal injection
To test whether use of the bs-AS-CK2 AS OGN would be effective to downregulate both CK2α and α′ proteins in vivo, we established localization of the OGN in mouse tissues and tumors following systemic administration. Athymic nude mice bearing orthotopic PC3-LN4 prostate tumors were given two intraperitoneal (i.p.) injections with FITC-labeled bs-AS-CK2 with a 24 h interval. Tissues were collected 24 h after the second injection, and the lysates were analyzed for FITC fluorescence. Background, calculated from the tissue lysates of mice processed equivalently but injected with PBS, was subtracted and dose ratios were calculated. FITC signal was detected at the highest levels in kidney and tumor, and was also detected in spleen, liver, blood, and heart (Fig. 3a). Verification of OGN localization in tumor was obtained by immunofluorescence analysis of the sectioned tumors using confocal microscopy. Processing of the tumor section with both an anti-FITC antibody to detect the OGN and an anti-CK8 antibody to mark tumor cells illustrated that FITC-labeled OGN was present in both the nuclear and cytosolic compartments of tumor cells at 24 h following the second dose (Fig. 3b).

a Biodistribution of FITC-bs-AS-CK2 in prostate tumor-bearing mice. Mice (n = 3) bearing orthotopic xenograft PC3-LN4 prostate tumors were injected i.p. with 2 doses of FITC-labeled bs-AS-CK2 (66 μg/kg) 24 h apart. Tissues were collected 24 h after the second dose, weighed, and homogenized in lysis buffer. FITC fluorescence was quantified against a standard curve. Background was subtracted from mice processed equivalently but treated with PBS and dose ratios were calculated as % injected dose per gram wet tissue weight. Data is presented as the mean ± SEM. b Localization of FITC-bs-AS-CK2 in tumor tissue. Representative mouse tissue from the biodistribution study illustrates that FITC-labeled OGN was present in tumor at 24 h following the second dose. Tumors were paraffin embedded and processed for confocal immunofluorescence microscopy using a double-label strategy of anti-FITC (green) and anti-CK8 (red) antibodies to mark OGN and tumor, respectively. A–C represents a section processed with primary antibodies and D–F represents a section processed without primary antibodies at ×600
Acute dosing regimen demonstrates no damage to normal tissues
We have previously published evidence of a potential pharmacologic window for inhibition or downregulation of CK2 expression in cancer cells compared to non-malignant cells. Injection of AS to CK2α directly into normal mouse prostate resulted in no abnormal histology and no significant induction of apoptosis [27]. Here we report on the effect of acute cumulative systemic dosing with bs-AS-CK2. Control and PC3-LN4 orthotopic xenograft prostate tumor-bearing mice were injected with bs-AS-CK2 or GAPDH OGNs at 2 dose levels or with saline. Intraperitoneal injections were given twice with a 24-h interval, and 10 days following the first injection tissues and tumor were processed for histology. Following injection of nontumor-bearing mice with 0.7 mg/kg bs-AS-CK2, no injury to prostate or liver tissues was observed by histologic examination (Fig. 4, panel a and data not shown). In contrast, injection of tumor-bearing mice with bs-AS-CK2 caused extensive death of tumor tissue cells (Fig. 4, panel b). Notably, an injection with a tenfold higher cumulative dose of bs-AS-CK2 than effectively induced cell death in tumor did not induce histologic change in the liver of nontumor-bearing mice (Fig. 4, panel c). Thus, there appears to be a pharmacologic window where malignant cells are more responsive to exposure to bs-AS-CK2 OGN as compared to non-malignant cells.

Acute dosing regimens with bs-AS-CK2 indicate no damage to normal tissues. Tissue from normal, non-tumor-bearing animals (a, c, e) or tumor-bearing (b, d, f) were processed for H&E staining 10 days following acute cumulative dosing of bs-AS-CK2, AS-GAPDH or saline given as two injections with a 24 h interval as labeled in the figure. Each treatment group included three mice. Images were captured at ×400
Dose response in vivo using unformulated bs-AS-CK2
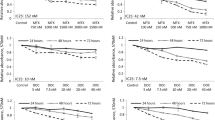
Initial experiments to establish a dose response of orthotopic prostate tumors in nude mice were carried out using bs-AS-CK2 PTO in saline. Orthotopic xenograft prostate tumors were established in male nude mice by the injection of PC3-LN4 cells directly into the prostate. After approximately 2 weeks the tumors were palpable, and the mice were divided into treatment groups. Mice received two i.p. injections of either bs-AS-CK2 or AS-GAPDH given with an interval of 24 h. Each injection comprised 50% of the cumulative dose. Ten to thirteen days following the first treatment, the tumor tissue was harvested and analyzed. In these and the subsequent experiments, AS to GAPDH was consistently used as a control OGN treatment, and all of the tumor weight data are presented relative to this control. As is shown in Fig. 5a, all dose amounts resulted in decreased tumor weight, with the best reduction in tumor weight observed at 132 μg/kg bs-AS-CK2 to 52% relative to the 132 μg/kg AS-GAPDH treatment. Interestingly, at this highest cumulative dose of bs-AS-CK2, a marked reduction in retro-peritoneal lymph node tumor volume was also observed (~13-fold reduction or 8% relative to 132 μg/kg AS-GAPDH treatment, data not shown).

Dose response of orthotopic prostate tumors following bs-AS-CK2 PTO treatment. Mice bearing orthotopic PC3-LN4 prostate tumors were treated i.p. with decreasing cumulative doses starting from 132 μg/kg of PTO, either AS-GAPDH or bs-AS-CK2. Mice were dosed twice with a 24 h interval. Mice were killed 10–13 days after starting treatment. a Tumor weight comparisons between different treatment groups. Results are reported as means ± SEM for group sizes of 3–8 animals. b Immunoblot analyses of fractionated tumor lysates. Representative signals from nuclear matrix of two tumors in each treatment groups are shown. Lamin B was used as a loading control
Quantitative real-time PCR analysis was performed following reverse transcription of the tumor RNA. The data shown in Table 2 demonstrate that at the three highest doses of bs-AS-CK2, both CK2α and α′ mRNA steady-state levels were reduced by approximately 50%. In the tumor treated with the lowest cumulative dose, CK2α, but not CK2α′, mRNA levels were reduced. AS-GAPDH treatment did not affect CK2 catalytic subunit mRNA levels relative to saline treatment. If the bs-AS-CK2 tumor sample mRNA levels were expressed relative to GAPDH, CK2α levels remain very similarly reduced, whereas CK2α′ levels are somewhat raised. Tumor tissue was also lysed and fractionated into nuclear matrix and cytosol for immunoblot analyses. Representative nuclear matrix fraction results for two tumors from each group are shown in Fig. 5b, whereas average expression data for all tumors is quantitated and shown in Table 3. Treatment with all dose amounts resulted in decreased CK2α and α′ steady-state protein levels, and the best reduction was observed at a cumulative dose of 33 μg/kg (Fig. 5b; Table 3). In one dose response experiment, a group of mice also received saline injections as a control, and these tumors were used as a further control for the dose response protein immunoblot data to examine whether injection of AS to GAPDH affected CK2 protein expression. As is shown in Table 3, CK2 protein expression levels in AS-GAPDH injected tumors were slightly reduced for CK2α, and raised for CK2α′ when expressed relative to saline; indicating that the reduction in CK2α and α′ protein steady-state levels is potentially slightly under- and over-represented when expressed relative to AS-GAPDH, respectively. CK2α and α′ steady-state protein levels were not appreciably reduced in the cytosolic fraction. AS treatment results in decreased steady-state gene expression at the protein level. Mechanistically this can occur through cleavage and loss of the mRNA transcript or by blockade of protein translation. Our data indicates that, following systemic treatment with bs-AS-CK2, loss of CK2α and α′ mRNA transcripts occurs and results in decreased protein steady-state levels in the nuclear compartment.
Tumor reduction is schedule sensitive
In the above-described dose response experiments, treatment with bs-AS-CK2 was given as two i.p. injections with a 24-h interval. A further question we posed was whether the tumor response to bs-AS-CK2 is schedule sensitive. Orthotopic xenograft prostate tumors were generated as described above for the dose response experiment, and once the tumors reached palpable detection, the mice were separated into groups of 5. We chose a cumulative dose amount of 66 μg/kg, and mice received this cumulative dose divided and given as an i.p. injection on different schedules as follows: 1 × 66 μg/kg given day 1; 2 × 33 μg/kg given days 1 and 2 (q1d); 2 × 33 μg/kg given days 1 and 4 (q4d); 4 × 16.5 μg/kg given days 1, 2, 3 and 4 (q1d). Thirteen days following the first treatment, the tumor tissue was harvested and analyzed. All treatment schedules resulted in decreased tumor weight relative to treatment with AS-GAPDH given 1 × 66 μg/kg (Fig. 6a). There was a markedly better primary tumor weight reduction to 26% of the control AS-GAPDH treatment observed in the mice receiving the cumulative dose on the 4 × 16.5 μg/kg schedule compared to the other schedules. This response is more than twofold improved over the best dose response results (Fig. 5a) in which twofold more drug was given cumulatively.

Tumor response to bs-AS-CK2 PTO treatment is schedule-dependent. Mice bearing orthotopic PC3-LN4 prostate tumors were treated i.p. with a single 66 μg/kg dose of AS-GAPDH PTO or with bs-AS-CK2 PTO using 1 of 4 different schedules: single dose of 66 μg/kg, 2 × 33 μg/kg doses spaced 1 d apart (q1d), 2 × 33 μg/kg doses spaced 4 d apart (q4d), or 4 × 16.5 μg/kg doses spaced 1 d apart (q1d). Mice were killed 13 days after starting treatment. a Tumor weight comparisons between different treatment groups. Results are reported as means ± SEM for group sizes of five animals. *Significant difference from control (P < 0.05). b Immunoblot analyses of fractionated tumor lysates. Representative signals from nuclear matrix or cytosol of two tumors in each treatment group are shown. The upper four blots represent nuclear matrix fraction, and the lower two blots represent cytosolic fraction. Lamin B and actin were used as loading controls for nuclear matrix and cytosol, respectively
Quantitative real-time PCR analysis was performed following reverse transcription of the tumor RNA. These data are shown in Table 4 and the CK2 mRNA levels are presented relative to AS-GAPDH treated tumors for consistency with the protein immunoblot data. Consistent with the dose response data, CK2α mRNA steady-state levels were reduced by all schedules; however, in contrast with the dose response data, CK2α′ mRNA levels showed increased expression for all schedules. This increased CK2α′ mRNA was observed when the data were analyzed relative to the saline samples from the dose response experiment as well. Tumor tissue was again lysed and fractionated into nuclear matrix and cytosol for immunoblot analyses. Representative results from the nuclear matrix fraction for two tumors from each group are shown in Fig. 6b, whereas average expression data for all tumors is quantitated and shown in Table 5. Again, treatment with all dose amounts resulted in decreased CK2α and α′ steady-state protein levels, with the best reduction observed using the 4 × 16.5 μg/kg schedule (Fig. 6b; Table 5). In contrast to the PCR data, the protein expression level data correlated well with the tumor weight data. We also examined the effects of bs-AS-CK2 treatment on CK2β and NF-κB p65 protein steady-state expression levels in the nuclear matrix fraction. Treatment on the 4 × 16.5 μg/kg schedule resulted in the best reduction in CK2β and NF-κB p65 protein (Fig. 6b; Table 5). In the cytosolic fraction, CK2α and α′ steady-state protein levels were reduced to ~80% of GAPDH levels in the 4 × 16.5 μg/kg schedule tumor samples. Interestingly, AKT steady-state protein levels in the cytosol were reduced in both the 4 × 16.5 and 2 × 33 μg/kg q4d treatments (Fig. 6b; Table 5). Phospho-serine 473 AKT protein expression was detected at similar levels to total AKT (Table 5). This data suggests that tumors from mice which received the drug more closely to the study termination date (i.e., day 4 vs. days 1 or 2) showed an AKT effect.
Discussion
In the present work, we observed an upregulation of CK2α′ in cancer cells after in vivo treatment of tumor bearing mice with AS OGN to CK2α. This suggests the possibility that even though CK2α′ is not essential for viability in most normal cells [68], its functionality in cancer cells might be altered so that it compensates for CK2α and conceivably promotes cell survival. We have described the characterization of an AS OGN with bispecific targeting of CK2α and α′ in both cultured cell and mouse xenograft models. In cell culture, bs-AS-CK2 effectively reduced CK2α and α′ protein expression, inhibited cell proliferation, and induced apoptosis. In mouse orthotopic xenograft PCa studies, systemic administration of the bs-AS-CK2 resulted in tumor localization of the OGN in both nuclear and cytoplasmic cellular compartments. The OGN also was detected in kidney, spleen, liver, blood, and heart. A similar normal tissue biodistribution pattern of PTO was reported after intravenous (i.v.) administration of 2′-O-(2-methoxyethyl)-ribose modified PTO against tumor necrosis factor-α [69]. Further, injection of much larger bs-AS-CK2 doses than used for the tumor response studies did not induce any detectable damage in normal tissue.
In our short-term tumor response studies, just one to four injections of bs-AS-CK2 PTO in dose ranges of 16.5–66 μg/kg (cumulative dose 66 μg/kg) resulted in significantly decreased tumor weight 13 days after the first injection. One difficult aspect of the use of orthotopic xenograft prostate tumors is the variable size of the tumors at treatment initiation. The PC3-LN4 cell line used here does not secrete PSA and is not labeled with a fluorescent or luminescent marker; thus, assessment of tumor response after treatment cannot be compared to an initial tumor size or other measured value. However, as a means of comparison, much larger cumulative doses are typically reported in studies examining the use of AS OGN to treat mouse xenograft tumors. For example, in a PCa study using i.p. injection of ATL1101 targeting insulin-like growth factor receptor (IGF-1R), representing PTO 2′-O-(2-methoxyethyl)-ribose gapmer chemistry, 50% reduction in tumor growth rate was obtained at a dose of 15 mg/kg given 25 times over 7 weeks (cumulative dose 375 mg/kg) [70]. In a bladder cancer study targeting survivin and human telomerase reverse transcriptase (hTERT) using partial PTO chemistry, treatment with 20 mg/kg was begun 1 day following tumor cell implantation and was given 9 times over 3 weeks (cumulative dose 180 mg/kg). Treatment with AS to survivin was effective at inhibiting tumor growth, and treatment with AS to hTERT was effective at reducing tumor growth [71]. In conclusion, our data indicate that molecular targeting of CK2 as an anti-cancer therapy strategy is very effective at lower doses than typically reported for other targets. Moreover, administration of the bs-AS-CK2 therapeutic resulted in reduced CK2α and α′ protein steady-state levels which correlated well with the effect of the treatment on tumor weight. Decreased steady-state mRNA levels for CK2α also reflected tumor weight response in a dose-dependent, but not strongly schedule-dependent, manner.
The best tumor weight reduction was obtained using the 4 × 16.5 μg/kg dosing schedule. We also observed the best combined reductions in protein steady-state levels for CK2α, CK2α′, CK2β, and NF-κB p65 using this same dosing schedule. These results suggest that sustained, lower dosing levels allowed for more comprehensive effects on CK2 holoenzyme expression at the protein level as well as on CK2-influenced signaling pathways such as NF-κB. Similar results were reported using the HNSCC model [55]. Given the prominent role played by NF-κB signaling in tumor initiating or cancer stem cell proliferation and in epithelial mesenchymal transition (EMT) and the metastasis process [72–75], it is particularly significant that downregulation of CK2 in these acute response studies also induced downregulation of nuclear NF-κB. AKT, also known as protein kinase B (PKB), is one of the most commonly activated signaling pathways in cancer [76]. Tissue culture experiments using established cell lines examining the effects of short-term CK2 inhibition on AKT have typically reported loss of AKT phosphorylation and activation status or activity, with little to no change in overall AKT protein level [50, 77]. A recent report using peripheral blood mononuclear cells (PBMCs) from chronic lymphocytic leukemia patients demonstrated that exposure to high doses of CK2 inhibitors led to decreased AKT protein levels which was preceded by loss of AKT phosphorylation at serine 473 [78]. Here, we observed a decrease in overall AKT protein levels and a parallel loss of phospho-serine 473 status 9 days after systemic exposure to bs-AS-CK2 in mice. Taken together, these data suggest that longer-term downregulation of CK2 also downregulated the protein expression of key cancer proliferative signaling pathway components.
In previous work, we observed that normal or benign cells in culture as well as normal prostate cells in vivo were relatively insensitive to CK2α AS OGN at concentrations that induced potent apoptosis in malignant cells [27]. Likewise, we observed no obvious injurious effects in normal mouse tissues in the present studies. However, despite the relative resistance of normal cells to downregulation of CK2, we believe that it would be important for future targeting of CK2 in cancer therapeutics to deliver a CK2 targeting agent in a way that is specifically directed to malignant cells while sparing the normal cells. This is due to the ubiquitous and cell survival properties of CK2 cellular functions and the related concerns of host toxicity with longer term treatment. In subsequent studies, we will report on the use of a bs-AS-CK2 OGN encapsulated within tenfibgen protein to simultaneously protect the OGN and direct it specifically to malignant cells.
Abbreviations
- AS:
-
Antisense
- bs:
-
Bispecific
- CK2:
-
Official acronym for former casein kinase 2 or II
- CRPC:
-
Castration-resistant prostate cancer
- d:
-
Day(s)
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- H&E:
-
Hematoxylin and eosin stain
- h:
-
Hour(s)
- HNSCC:
-
Head and neck squamous cell carcinoma
- i.p.:
-
Intraperitoneal
- i.v.:
-
Intravenous
- kDa:
-
Kilo Dalton
- LDH:
-
Lactate dehydrogenase
- min:
-
Minutes
- NF-κB:
-
Nuclear factor kappa B
- OGN:
-
Oligonucleotide
- PBS:
-
Phosphate-buffered saline
- PCa:
-
Prostate cancer
- PCR:
-
Polymerase chain reaction
- PKB:
-
Protein kinase B
- PTO:
-
Phosphorothioate-modified oligodeoxynucleotide
- RFP:
-
Red fluorescent protein
- s:
-
Seconds
- Ser:
-
Serine
References
Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2–a key suppressor of apoptosis. Adv Enzyme Regul 48:179–187. doi:10.1016/j.advenzreg.2008.04.002
Pinna LA, Allende JE (2009) Protein kinase CK2 in health and disease: Protein kinase CK2: an ugly duckling in the kinome pond. Cell Mol Life Sci 66(11–12):1795–1799. doi:10.1007/s00018-009-9148-9
Guerra B, Issinger OG (2008) Protein kinase CK2 in human diseases. Curr Med Chem 15(19):1870–1886. doi:10.1007/s00018-009-9148-9
Trembley JH, Wang G, Unger G, Slaton J, Ahmed K (2009) Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci 66(11–12):1858–1867. doi:10.1007/s00018-009-9154-y
Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, Ahmed K (2010) Emergence of protein kinase CK2 as a key target in cancer therapy. BioFactors 36(3):187–195. doi:10.1002/biof.96
Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA (2009) Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta 1793(5):847–859. doi:10.1016/j.bbamcr.2009.01.013
Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17(3):349–368. doi:10.1096/fj.02-0473rev
Graham KC, Litchfield DW (2000) The regulatory β subunit of protein kinase CK2 mediates formation of tetrameric CK2 complexes. J Biol Chem 275(7):5003–5010. doi:10.1074/jbc.275.7.5003
Zhang P, Davis AT, Ahmed K (1998) Mechanism of protein kinase CK2 association with nuclear matrix: role of disulfide bond formation. J Cell Biochem 69(2):211–220. doi:10.1002/(SICI)1097-4644(19980501)69:2<211:AID-JCB11>3.0.CO;2-H
Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K (2001) Protein kinase CK2 signal in neoplasia. Histol Histopathol 16(2):573–582
Ahmed K, Davis AT, Wang H, Faust RA, Yu S, Tawfic S (2000) Significance of protein kinase CK2 nuclear signaling in neoplasia. J Cell Biochem Suppl 35:130–135. doi:10.1002/1097-4644(2000)79:35+<130:AID-JCB1136>3.0.CO;2-N
Ahmed K (1999) Nuclear matrix and protein kinase CK2 signaling. Crit Rev Eukaryot Gene Expr 9(3–4):329–336
St-Denis NA, Litchfield DW (2009) Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci 66(11–12):1817–1829. doi:10.1007/s00018-009-9150-2
Ahmed K (1971) Studies on nuclear phosphoproteins of rat ventral prostate: incorporation of 32P from γ-32P-ATP. Biochim Biophys Acta 243:38–48
Ahmed K, Ishida H (1971) Effect of testosterone on nuclear phosphoproteins of rat ventral prostate. Mol Pharmacol 7(3):323–327
Guo C, Yu S, Davis AT, Ahmed K (1999) Nuclear matrix targeting of the protein kinase CK2 signal as a common downstream response to androgen or growth factor stimulation of prostate cancer cells. Cancer Res 59(5):1146–1151
Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K (2001) A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem 276(8):5992–5999. doi:10.1074/jbc.M004862200
Tawfic S, Faust RA, Gapany M, Ahmed K (1996) Nuclear matrix as an anchor for protein kinase CK2 nuclear signalling. J Cell Biochem 62(2):165–171. doi:10.1002/(SICI)1097-4644(199608)62:2<165:AID-JCB4>3.0.CO;2-Q
Ahmed K, Yenice S, Davis A, Goueli SA (1993) Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc Natl Acad Sci USA 90(10):4426–4430
Tawfic S, Goueli SA, Olson MO, Ahmed K (1994) Androgenic regulation of phosphorylation and stability of nucleolar protein nucleolin in rat ventral prostate. Prostate 24(2):101–106
Tawfic S, Ahmed K (1994) Growth stimulus-mediated differential translocation of casein kinase 2 to the nuclear matrix. Evidence based on androgen action in the prostate. J Biol Chem 269(40):24615–24620
Yu S, Davis AT, Guo C, Green JE, Ahmed K (1999) Differential targeting of protein kinase CK2 to the nuclear matrix upon transient overexpression of its subunits. J Cell Biochem 74(1):127–134. doi:10.1002/(SICI)1097-4644(19990701)74:1<127:AID-JCB14>3.0.CO;2-3
Guo C, Davis AT, Ahmed K (1998) Dynamics of protein kinase CK2 association with nucleosomes in relation to transcriptional activity. J Biol Chem 273(22):13675–13680. doi:10.1074/jbc.273.22.13675
Yu S, Wang H, Davis A, Ahmed K (2001) Consequences of CK2 signaling to the nuclear matrix. Mol Cell Biochem 227(1–2):67–71. doi:10.1023/A:1013156721938
Elcock LS, Bridger JM (2008) Exploring the effects of a dysfunctional nuclear matrix. Biochem Soc Trans 36(Pt 6):1378–1383. doi:10.1042/BST0361378
Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H (2004) PML nuclear bodies and apoptosis. Oncogene 23(16):2819–2824. doi:10.1038/sj.onc.1207533
Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K (2004) Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res 2(12):712–721
Wang H, Davis A, Yu S, Ahmed K (2001) Response of cancer cells to molecular interruption of the CK2 signal. Mol Cell Biochem 227(1–2):167–174. doi:10.1023/A:1013112908734
Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K (2005) Targeting CK2 for cancer therapy. Anticancer Drugs 16(10):1037–1043. doi:10.1097/00001813-200511000-00001
Rayan A, Goueli SA, Lange P, Ahmed K (1985) Chromatin-associated protein kinases in human normal and benign hyperplastic prostate. Cancer Res 45(5):2277–2282
Yenice S, Davis AT, Goueli SA, Akdas A, Limas C, Ahmed K (1994) Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 24(1):11–16
Ruzzene M, Pinna LA (2010) Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta 1804(3):499–504. doi:10.1016/j.bbapap.2009.07.018
Faust RA, Niehans G, Gapany M, Hoistad D, Knapp D, Cherwitz D, Davis A, Adams GL, Ahmed K (1999) Subcellular immunolocalization of protein kinase CK2 in normal and carcinoma cells. Int J Biochem Cell Biol 31(9):941–949. doi:10.1016/S1357-2725(99)00050-3
Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K (1996) Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett 101(1):31–35. doi:10.1016/0304-3835(96)04110-9
Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K (1995) Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med 1(6):659–666
Giusiano S, Cochet C, Filhol O, Duchemin-Pelletier E, Secq V, Bonnier P, Carcopino X, Boubli L, Birnbaum D, Garcia S, Iovanna J, Charpin C (2010) Protein kinase CK2alpha subunit over-expression correlates with metastatic risk in breast carcinomas: quantitative immunohistochemistry in tissue microarrays. Eur J Cancer. doi:10.1016/j.ejca.2010.11.028
Laramas M, Pasquier D, Filhol O, Ringeisen F, Descotes JL, Cochet C (2007) Nuclear localization of protein kinase CK2 catalytic subunit (CK2α) is associated with poor prognostic factors in human prostate cancer. Eur J Cancer 43(5):928–934. doi:10.1016/j.ejca.2006.11.021
O-charoenrat P, Rusch V, Talbot SG, Sarkaria I, Viale A, Socci N, Ngai I, Rao P, Singh B (2004) Casein kinase II alpha subunit and C1-inhibitor are independent predictors of outcome in patients with squamous cell carcinoma of the lung. Clin Cancer Res 10(17):5792–5803. doi:10.1158/1078-0432.CCR-03-0317
Kim JS, Eom JI, Cheong J-W, Choi AJ, Lee JK, Yang WI, Min YH (2007) Protein kinase CK2α as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res 13(3):1019–1028. doi:10.1158/1078-0432.ccr-06-1602
Lin KY, Fang CL, Chen Y, Li CF, Chen SH, Kuo CY, Tai C, Uen YH (2010) Overexpression of nuclear protein kinase CK2 Beta subunit and prognosis in human gastric carcinoma. Ann Surg Oncol 17(6):1695–1702. doi:10.1245/s10434-010-0911-9
Lin KY, Tai C, Hsu JC, Li CF, Fang CL, Lai HC, Hseu YC, Lin YF, Uen YH (2011) Overexpression of nuclear protein kinase CK2 alpha catalytic subunit (CK2alpha) as a poor prognosticator in human colorectal cancer. PLoS One 6(2):e17193. doi:10.1371/journal.pone.0017193
McKenzie S, Kyprianou N (2006) Apoptosis evasion: the role of survival pathways in prostate cancer progression and therapeutic resistance. J Cell Biochem 97(1):18–32. doi:10.1002/jcb.20634
Ahmed K, Gerber DA, Cochet C (2002) Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol 12(5):226–230. doi:10.1016/S0962-8924(02)02279-1
Kelliher MA, Seldin DC, Leder P (1996) Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIα. EMBO J 15(19):5160–5166
Landesman-Bollag E, Channavajhala PL, Cardiff RD, Seldin DC (1998) p53 deficiency and misexpression of protein kinase CK2α collaborate in the development of thymic lymphomas in mice. Oncogene 16(23):2965–2974. doi:10.1038/sj.onc.1201854
Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC (2001) Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 20(25):3247–3257. doi:10.1038/sj.onc.1204411
Landesman-Bollag E, Song DH, Romieu-Mourez R, Sussman DJ, Cardiff RD, Sonenshein GE, Seldin DC (2001) Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem 227(1–2):153–165. doi:10.1023/A:1013108822847
Channavajhala P, Seldin DC (2002) Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 21(34):5280–5288. doi:10.1038/sj.onc.1205640
Hung MS, Lin YC, Mao JH, Kim IJ, Xu Z, Yang CT, Jablons DM, You L (2010) Functional polymorphism of the CK2alpha intronless gene plays oncogenic roles in lung cancer. PLoS One 5(7):e11418. doi:10.1371/journal.pone.0011418
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K (2010) CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 70(24):10288–10298. doi:10.1158/0008-5472.can-10-1893
Perea SE, Reyes O, Puchades Y, Mendoza O, Vispo NS, Torrens I, Santos A, Silva R, Acevedo B, Lopez E, Falcon V, Alonso DF (2004) Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res 64(19):7127–7129. doi:10.1158/0008-5472.CAN-04-2086
Perea SE, Reyes O, Baladron I, Perera Y, Farina H, Gil J, Rodriguez A, Bacardi D, Marcelo JL, Cosme K, Cruz M, Valenzuela C, Lopez-Saura PA, Puchades Y, Serrano JM, Mendoza O, Castellanos L, Sanchez A, Betancourt L, Besada V, Silva R, Lopez E, Falcon V, Hernandez I, Solares M, Santana A, Diaz A, Ramos T, Lopez C, Ariosa J, Gonzalez LJ, Garay H, Gomez D, Gomez R, Alonso DF, Sigman H, Herrera L, Acevedo B (2008) CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol Cell Biochem 316(1–2):163–167. doi:10.1007/s11010-008-9814-5
Perera Y, Farina HG, Hernandez I, Mendoza O, Serrano JM, Reyes O, Gomez DE, Gomez RE, Acevedo BE, Alonso DF, Perea SE (2008) Systemic administration of a peptide that impairs the protein kinase (CK2) phosphorylation reduces solid tumor growth in mice. Int J Cancer 122(1):57–62. doi:10.1002/ijc.23013
Solares AM, Santana A, Baladron I, Valenzuela C, Gonzalez CA, Diaz A, Castillo D, Ramos T, Gomez R, Alonso DF, Herrera L, Sigman H, Perea SE, Acevedo BE, Lopez-Saura P (2009) Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 9:146. doi:10.1186/1471-2407-9-146
Brown MS, Diallo OT, Hu M, Ehsanian R, Yang X, Arun P, Lu H, Korman V, Unger G, Ahmed K, Van Waes C, Chen Z (2010) CK2 modulation of NF-κB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin Cancer Res 16(8):2295–2307. doi:10.1158/1078-0432.ccr-09-3200
Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, Fidler IJ (1996) Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res 2(9):1627–1636
Hayward S, Dahiya R, Cunha G, Bartek J, Deshpande N, Narayan P (1995) Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell Dev Biol Anim 31(1):14–24. doi:10.1007/BF02631333
Yamane K, Kinsella TJ (2005) CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res 65(10):4362–4367. doi:10.1158/0008-5472.CAN-04-3941
Ulloa L, Diaz-Nido J, Avila J (1993) Depletion of casein kinase II by antisense oligonucleotide prevents neuritogenesis in neuroblastoma cells. EMBO J 12(4):1633–1640
Fukuhara Y, Takeshima T, Kashiwaya Y, Shimoda K, Ishitani R, Nakashima K (2001) GAPDH knockdown rescues mesencephalic dopaminergic neurons from MPP+-induced apoptosis. Neuroreport 12(9):2049–2052
Gaur U, Sahoo SK, De TK, Ghosh PC, Maitra A, Ghosh PK (2000) Biodistribution of fluoresceinated dextran using novel nanoparticles evading reticuloendothelial system. Int J Pharm 202(1–2):1–10. doi:10.1016/S0378-5173(99)00447-0
Son YJ, Jang JS, Cho YW, Chung H, Park RW, Kwon IC, Kim IS, Park JY, Seo SB, Park CR, Jeong SY (2003) Biodistribution and anti-tumor efficacy of doxorubicin loaded glycol-chitosan nanoaggregates by EPR effect. J Control Release 91(1–2):135–145. doi:10.1016/S0168-3659(03)00231-1
Ahmed RL, Davis AT, Ahmed K (1996) Interference in protein assays of biological specimens by vanadyl compounds. Anal Biochem 237(1):76–79. doi:10.1006/abio.1996.0203
Goueli SA, Hanten J, Davis A, Ahmed K (1990) Polyclonal antibodies against rat liver cytosolic casein kinase II (CK-2) cross-react with CK-2 from other tissues and nuclear form (PK-N2) of the enzyme. Biochem Int 21(4):685–694
Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW (1979) Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 17(1):16–23
Kozlowski JM, Fidler IJ, Campbell D, Xu Z-l, Kaighn ME, Hart IR (1984) Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res 44(8):3522–3529
Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR (2001) Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res 61(22):8135–8142
Seldin DC, Lou DY, Toselli P, Landesman-Bollag E, Dominguez I (2008) Gene targeting of CK2 catalytic subunits. Mol Cell Biochem 316(1–2):141–147. doi:10.1007/s11010-008-9811-8
Geary RS, Yu RZ, Watanabe T, Henry SP, Hardee GE, Chappell A, Matson J, Sasmor H, Cummins L, Levin AA (2003) Pharmacokinetics of a tumor necrosis factor-α phosphorothioate 2’-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug Metab Dispos 31(11):1419–1428. doi:10.1124/dmd.31.11.1419
Furukawa J, Wraight CJ, Freier SM, Peralta E, Atley LM, Monia BP, Gleave ME, Cox ME (2010) Antisense oligonucleotide targeting of insulin-like growth factor-1 receptor (IGF-1R) in prostate cancer. Prostate 70(2):206–218. doi:10.1002/pros.21054
Kunze D, Wuttig D, Kausch I, Blietz C, Blumhoff L, Burmeister Y, Kraemer K, Fuessel S, Toma M, Schwenzer B, Meye A, Grimm MO, Hakenberg OW, Jocham D, Wirth MP (2008) Antisense-mediated inhibition of survivin, hTERT and VEGF in bladder cancer cells in vitro and in vivo. Int J Oncol 32(5):1049–1056
Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI (2011) Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun 2(1):162. doi:10.1038/ncomms1159
Sethi S, Macoska J, Chen W, Sarkar FH (2010) Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res 3(1):90–99
Zhang Q, Helfand BT, Jang TL, Zhu LJ, Chen L, Yang XJ, Kozlowski J, Smith N, Kundu SD, Yang G, Raji AA, Javonovic B, Pins M, Lindholm P, Guo Y, Catalona WJ, Lee C (2009) Nuclear factor-κB-mediated transforming growth factor-β-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res 15(10):3557–3567. doi:10.1158/1078-0432.ccr-08-1656
Min C, Eddy SF, Sherr DH, Sonenshein GE (2008) NF-κB and epithelial to mesenchymal transition of cancer. J Cell Biochem 104(3):733–744. doi:10.1002/jcb.21695
Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8(8):627–644. doi:10.1038/nrd2926
Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F, Pinna LA, Ruzzene M (2005) Protein kinase CK2 phosphorylates and upregulates Akt//PKB. Cell Death Differ 12(6):668–677
Shehata M, Schnabl S, Demirtas D, Hilgarth M, Hubmann R, Ponath E, Badrnya S, Lehner C, Hoelbl A, Duechler M, Gaiger A, Zielinski C, Schwarzmeier JD, Jaeger U (2010) Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood 116(14):2513–2521. doi:10.1182/blood-2009-10-248054
Acknowledgments
The authors thank Ms. Lindsey M. Watch for excellent technical assistance. Grant Sponsors: Department of Veterans Affairs Medical Research Merit Review Funds (KA); National Cancer Institute grant numbers UO1-CA15062 (KA) and RO1-CA150182 (KA); National Institute of Health grant number R01-DK067436 (BK).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Trembley, J.H., Unger, G.M., Tobolt, D.K. et al. Systemic administration of antisense oligonucleotides simultaneously targeting CK2α and α′ subunits reduces orthotopic xenograft prostate tumors in mice. Mol Cell Biochem 356, 21–35 (2011). https://doi.org/10.1007/s11010-011-0943-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-011-0943-x