
Abstract
Nobiletin, a compound isolated from citrus fruits, is a polymethoxylated flavone derivative shown to have anti-inflammatory, antitumor, and neuroprotective properties. This study has investigated that nobiletin exerted inhibitory effects on the cell adhesion, invasion, and migration abilities of a highly metastatic AGS cells under non-cytotoxic concentrations. Data also showed nobiletin could inhibit the activation of focal adhesion kinase (FAK) and phosphoinositide-3-kinase/Akt (PI3K/Akt) involved in the downregulation of the enzyme activities, protein expressions, messenger RNA levels of matrix metalloproteinase-2 (MMP-2), and matrix metalloproteinase-2 (MMP-9). Also, our data revealed that nobiletin inhibited FAK/PI3K/Akt with concurrent reduction in the protein expressions of Ras, c-Raf, Rac-1, Cdc42, and RhoA by western blotting, whereas the protein level of RhoB increased progressively. Otherwise, nobiletin-treated AGS cells showed tremendously decreased in the phosphorylation and degradation of inhibitor of kappaBα (IκBα), the nuclear level of NF-κB, and the binding ability of NF-κB to NF-κB response element. Furthermore, nobiletin significantly decreased the levels of phospho-Akt and MMP-2/9 in Akt1-cDNA-transfected cells concomitantly with a marked reduction in cell invasion and migration. These results suggest that nobiletin can reduce invasion and migration of AGS cells, and such a characteristic may be of great value in the development of a potential cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is a kind of gastrointestinal tract cancer which is the leading cause of cancer-related mortality in the world. Moreover, approximately 90% of gastric cancers are adenocarcinomas [1]. Studies have shown that a high intake of smoked, salted, nitrated foods, and carbohydrates, with a low intake of vegetables, fruits, and milks, are linked to cancer incidence. These diets have been shown to significantly increase the risk for gastric cancer [2]. Despite recent advances in gastric cancer treatment, there is still no effective cure for patients with advanced stages of this disease [1]. In recent years, attention has been focused on the anticancer properties of edible plants, which play an important role in the prevention of disease. Nobiletin (3′,4′,5,6,7,8-Hexamethoxyflavone) (Fig. 1a) is a major component in citrus fruits, particularly in the peels of sweet oranges (Citrus sinensis) and mandarin oranges (Citrus reticulate) [3, 4]. Previous papers have reported that nobiletin exhibits biological effects including anti-inflammatory, antitumor, and neuroprotective properties [5–7]. Kawabata et al. [8] reported that nobiletin has a distinct ability to highly suppress MMP-7 expression and production presumably by blocking AP-1 activity. In addition, it has been reported that nobiletin directly inhibits MEK activity and decreases the sequential phosphorylation of ERK, exhibiting antitumor activity by suppressing MMP expression in HT-1080 cells [9].

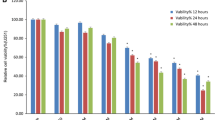
The effects of nobiletin on the viability in four cell lines, Chang liver, HepG2, Caco-2, and AGS cells. a Chemical structure of nobiletin. b Cultured cells were treated with or without nobiletin under various concentrations for 24 and 48 h. Thereafter, cell viabilities were determined by MTT assay. The survival cell number was directly proportional to formazan, which was measured spectrophotometrically at 563 nm. Values represent mean ± SD of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 compared with the untreated control (dose 0)
FAK is a nonreceptor tyrosine kinase primarily localized to cell–matrix adhesions which acts as a central regulator of focal adhesion to influence cell survival, differentiation, proliferation, metastasis, and tissue remodeling [10–12]. Metastasis of cancer cells involves multistep processes and various cytophysiological changes, including changed adhesion ability between the cells and the extracellular matrix (ECM) and damaged intercellular interaction. Excess ECM degradation is one of the hallmarks of tumor invasion and migration. Metastasis involves an overexpression of proteolytic enzymes, such as matrix metalloproteinases (MMPs). MMP-2 and MMP-9 (also known as type IV collagenases or gelatinases) can degrade most of the ECM components which form the basal membrane [13].
The Rho family of small guanosine triphosphatases (GTPases), which include RhoA, RhoB, c-Raf, Rac-1, and Cdc42, are critical in regulating actin reorganization associated with cell growth, migration, transformation, and gene expression [14]. The Rho family of GTPases has also been reported to be involved in the regulation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway [15]. In addition, Shih et al. [16] reported that PI3K/Akt signal transduction pathway regulates cell invasion and metastasis of non-small cell lung cancer (NSCLC) and is closely associated with the development and progression of various tumors.
NF-κB is a multisubunit transcription factor, which is involved in immune response, inflammation, and malignant transformation. Active NF-κB consists of p50, p52, p65 (RelA), Rel B, and c-Rel. Under normal conditions, NF-κB is maintained in the cytoplasm through interactions with an inhibitor of NF-κB (IκB), but upon dissociation, it moves into the nucleus and promotes the proliferation, angiogenesis and metastasis of cancer cells [17]. Previous studies have showed that MMP-2 and MMP-9 promoters are coordinately regulated by NF-κB [18, 19]. NF-κB regulates the expressions of matrix metalloproteinases, which is consistent with a role for this protein in regulating metastasis.
Although it is quite clear that nobiletin may inhibit the growth of various cancers by inducing cancer cells toward apoptosis, the precise impact of nobiletin on cancer invasion and migration is still uncertain. Thus, we investigated the effects and mechanisms by which nobiletin suppresses the invasion and migration of AGS cells.
Materials and methods
Reagents and antibodies
Nobiletin (Purity >99%) was purchased from Extrasynthese (Genay, France.); DMSO, Tris–HCl, EDTA, SDS, phenylmethylsulfonyl fluoride, bovine serum albumin (BSA), gelatin, crystal violet, leupeptin, Nonidet P-40, deoxycholic acid, and sodium orthovanadate were purchased from Sigma-Aldrich (St. Louis, MO, USA); the protein assay kit was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Dulbecco’s phosphate buffer solution (PBS), trypsin–EDTA, Dulbecco’s modified Eagle’s medium (DMEM), and RPMI medium 1640 were purchased from Life Technologies, Inc. (Gibco/BRL, Gaithersburg, MD). Antibody against IκBα, FAK, and PKB/Akt, proteins and phosphorylated proteins were purchased from Cell Signaling Tech. (Beverly, MA, USA). PI3K, MMP-2, MMP-9, Ras, c-Raf, Rac-1, Cdc42, RhoA, RhoB, NF-κB (p65 and p50), β-actin, C23 antibodies were purchased from BD Transduction Laboratories (San Jose, CA, USA). The enhanced chemiluminescence (ECL) kit was purchased from Amersham GE Healthcare UK Ltd. (Buckinghamshire, England).
Cell culture and nobiletin treatment
Human nonmalignant Chang liver cells, HepG2 hepatoblastoma cells, and Caco-2 colon adenocarcinoma cells were maintained in a DMEM. Human AGS gastric adenocarcinoma cell line was maintained in a RPMI-1640 medium. All cells were cultured at 37°C in a humidified atmosphere of 5% CO2–95% air. The culture medium was renewed every 2–3 days in medium supplemented with 10% fetal calf serum and antibiotics (100 U/ml of penicillin and 100 mg/ml of streptomycin). Adherent cells were detached by incubation with trypsin. For nobiletin treatment, the stock solution of nobiletin was dissolved in dimethylsulfoxide (DMSO) and sterilized by filtration through 0.2 μm disc filters. Suitable amounts of stock solution (1 mg/ml in DMSO) of nobiletin were added into the cultured medium to achieve the indicated concentrations (final DMSO concentration was <0.2%).
Assessment of cell viability (MTT assay)
To measure the effect of nobiletin on cell viability, the Chang Liver, HepG2, Caco-2, and AGS cells were seeded in 24-well plates (1 × 105 cells/well) for 16–18 h. Then, the cells were treated with or without nobiletin under various concentration (0, 1, 1.5, 2, 2.5, 3, 3.5, 4, and 4.5 μM) for various periods of time (24 and 48 h). At the end of the assay period, cell viability was measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide] assay, as described previously [20]. Each concentration was repeated three times. After the exposure period, the medium was removed and followed by washing of the cells with PBS. Then, the medium was changed and incubated with MTT solution (5 mg/ml)/well for 4 h. The medium was removed, and formazan was solubilized in isopropanol and measured spectrophotometrically at 563 nm. The percentage of viable cells was estimated by comparing with the untreated control cells.
Cell–matrix adhesion assay
First, the 24-well plate was coated with 500 μl type IV collagen (10 μg/ml) at 4°C overnight and then treated cells were seeded at a density of 1 × 105 cells/well for 30 min. Then, non-adherent cells were removed by PBS washes, and adherent cells were fixed in ethanol. After a staining with 0.1% crystal violet, fixed cells were lysed in 0.2% Triton-100, and measured spectrophotometrically at 550 nm.
Immunofluorescence staining assay
To determine the effect of nobiletin on cell morphology and actin stress fibers, AGS cells (4 × 105 cells/well) were plated in six-well plates and grown for 16–18 h and then incubated in the different concentrations of nobiletin (0, 1, 1.5, and 2 μM) for 24 h. After the exposure period, the media was removed, and the cells were washed with Ca2+/Mg2+ free PBS. Cells were then fixed with 4% paraformaldehyde in Ca2+/Mg2+ free PBS for 15 min and incubated with 0.5% Triton X-100/in Ca2+/Mg2+ free PBS for 5 min. Cells were incubated with 1% bovine serum albumin in 0.5% Triton X-100/in Ca2+/Mg2+ free PBS for 1 h (blocking) and then with 200 U/ml Alexa flour 488-phallodin (Invitrogen, Karlsruhe, Germany) for 1 h to stain the actin filaments. The fluorescent images were visualized with a BX51 fluorescence microscope (Olympus, Tokyo, Japan).
Boyden chamber invasion assay
The ability of AGS cells to pass through filters coated with Matrigel (BD Transduction Laboratories, San Jose, CA) was measured by Boyden chamber invasion assay [21]. Matrigel was diluted to 200 μg/ml with cold filtered distilled water and applied to the top side of a 8 μm pore polycarbonate filter. Briefly, AGS cells were treated with various concentrations of nobiletin for 48 h. After 48 h, cells were detached by trypsin and resuspended in serum-free medium. Medium containing 10% fetal bovine serum medium was applied to the lower chamber as chemoattractant, and then cells were then seeded on the upper chamber at a density of 1 × 105 cells/well in 50 μl of serum-free medium. The chamber was incubated for 8 h at 37°C. At the end of incubation, the cells in the upper surface of the membrane were carefully removed with a cotton swab. Cells invading across the Matrigel to the lower surface of the membrane were fixed with methanol and stained with 5% Giemsa solution. The invading cells on the lower surface of the membrane filter were counted with a light microscope. The data are presented as the average number of cells attached to the bottom surface from five random fields. Each experiment was carried out in triplicate.
Wound-healing assay
To determine cell motility determination, AGS cells (1 × 105cells/ml) were seeded in 6-well tissue culture plate and grown to 80–90% confluence. After aspirating the medium, the center of the cell monolayers was scraped with a sterile micropipette tip to create a denuded zone (gap) of constant width. Subsequently, cellular debris was washed with PBS, and the AGS cells were exposed to various concentrations of nobiletin (0, 1, 1.5, and 2 μM). The wound closure was monitored and photographed at 0, 12, 24, and 48 h with an Olympus CKX-41 inverted microscope and an Olympus E-410 camera. To quantify the migrated cells, pictures of the initial wounded monolayers were compared with the corresponding pictures of cells at the end incubation. Artificial lines fitting the cutting edges were drawn on pictures of the original wounds and overlaid on the pictures of cultures after incubation. Migrated cells across the white lines were counted in six random fields from each triplicate treatment, and data are presented as mean ± SD.
Analysis of MMP-2 and MMP-9 activities by zymography
Cells (4 × 105 cells/ml) were seeded into the 10-cm culture dish and then incubated in the different concentrations of nobiletin (0, 1, 1.5, and 2 μM) for 24 h. Additional groups of cells were treated with nobiletin at a concentration of 2 μM for different periods of times (0, 12, 24, 36, and 48 h). Subsequently, the conditioned medium was collected and gelatin zymography was performed to examine the activities of MMP-2 and MMP-9. Samples were mixed with loading buffer and electrophoresed on 8% SDS-polyacrylamide gel containing 0.1% gelatin. Electrophoresis was performed at 140 and 110 V for 3 h. Gels were then washed twice in zymography washing buffer (2.5% Triton X-100 in double-distilled H2O) at room temperature to remove SDS, followed by incubation at 37°C for 12–16 h in zymography reaction buffer (40 mM Tris–HCl, 10 mM CaCl2, 0.02% NaN3), stained with Coomassie blue R-250 (0.125% Coomassie blue R-250, 0.1% amino black, 50% methanol, 10% acetic acid) for 1 h and destained with destaining solution (20% methanol, 10% acetic acid, 70% double-distilled H2O). Nonstaining bands representing the levels of the latent forms of MMP-2 and MMP-9 were quantified by densitometer measurement using a digital imaging analysis system.
Isolation of total RNA, reverse transcriptase polymerase chain reaction (RT-PCR), and DNA electrophoresis
Total RNA was isolated from AGS cells using the total RNA Extraction Midiprep System (Viogene BioTek, Taiwan). Total RNA (2 μg) was transcribed to 20 μl cDNA with 1 μl deoxynucleotide triphosphate (dNTP; dNTP set consists of 2.5 mM aqueous solutions at pH 7.0 of each of dATP, dCTP, dGTP, and dTTP), 1 μl Oligo dT (10 pmol/ml), 1 μl RTase (200 U), 1 μl RNase inhibitor and 5× reaction buffer. The appropriate primers (sense of MMP-2, 5′-GGCCCTGTCACTCCTGAGAT-3′, nt 1337–1356; antisense of MMP-2, 5′-GGCATCCAGGTTATCGGGGA-3′, nt 2026–2007; sense of MMP-9, 5′-AGGCCTCTACAGAGTCTTTG-3′, nt 1201–1220; antisense of MMP-9, 5′-CAGTCCAACAAGAAAGGACG-3′, nt 1702–1683; sense of GADPH, 5′-CGGAGTCAACGGATTGGTGTT-3′ nt 94–126; antisense of GADPH, 5′-AGCCTTCTCCATGGTTGGTGAAGAC-3′, nt 399–375) were used for polymerase chain reaction (PCR) amplifications. PCR was performed with Platinum Taq polymerase (Invitrogen, San Diego, CA, USA) under the following conditions: 30 cycles of 94°C for 1 min, 59°C (MMP-2) or 60°C (MMP-9 and GAPDH) for 1 min, 72°C for 1 min followed by 10 min at 72°C. The final products were electrophoresed on 2% agarose gel and detected by ethidium bromide staining.
Preparation of whole-cell lysates and nuclear extracts
The cells were lysed with ice-cold RIPA buffer (1% NP-40, 50 mM Tris-base, 0.1% SDS, 0.5% deoxycholic acid, 150 mM NaCl, pH 7.5) and then the following inhibitors were added: phenylmethylsulfonyl fluoride (100 mM), leupeptin (100 mM), and sodium orthovanadate (100 mM). After vortexing for 30 min on ice, the samples were centrifuged at 12000×g for 10 min, and then the supernatants were collected, denatured, and subjected to SDS-PAGE and western blotting. Nuclear extracts were prepared as previously described [22]. Each nuclear pellet was resuspended in nuclear extract buffer (1.5 mM MgCl2, 10 mM HEPES, pH 7.9, 0.1 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 25% glycerol, and 420 mM NaCl). The nuclear suspension was incubated on ice for 20 min and then centrifuged at 14,000×g for 5 min. The supernatant (corresponding to the soluble nuclear fraction) was saved, and then used for NF-κB (p65 and p50) detection. The protein content was determined with Bio-Rad protein assay reagent using bovine serum albumin as a standard.
Western blotting analysis
To analyze the migration-related proteins, western blotting was performed as follows. The denatured samples (50 μg extracted protein) were resolved on 10–12% SDS-PAGE gels. The proteins were then transferred onto nitrocellulose membranes. Non-specific binding of the membranes was blocked with Tris-buffered saline (TBS) containing 1% (w/v) nonfat dry milk and 0.1% (v/v) Tween-20 (TBST) for more than 2 h. Membranes were washed with TBST three times for 10 min and incubated with an appropriate dilution of specific primary antibodies in TBST overnight at 4°C. Subsequently, membranes were washed with TBST and incubated with appropriate secondary antibodies (horseradish peroxidase-conjugated goat antimouse or antirabbit IgG) for 1 h. After washing the membranes three times for 10 min in TBST, the band detections were revealed by enhanced chemiluminescence using ECL western blotting detection reagents and exposed ECL hyperfilm in FUJFILM Las-3000 mini (Tokyo, Japan). Then proteins were quantitatively determined by densitometry using FUJFILM-Multi Gauge V3.0 software.
Analysis of NF-κB binding activity (electrophoretic mobility shift assay)
Cell nuclear proteins were extracted with a nuclear extract buffer and measured by electrophoretic mobility shift assay (EMSA) [23]. Cells (1 × 105/ml) were collected in PBS buffer (pH 7.4) and centrifuged at 2000×g for 5 min at 4°C. Cells were lysed with buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 0.5 mM PMSF (pH 7.9) containing 5% NP-40) for 10 min on ice, and this was followed by vortexing to shear the cytoplasmic membranes. The lysates were centrifuged at 2000×g for 10 min at 4°C. The pellet containing the nuclei was extracted with high salt buffer B (20 mM HEPES, 420 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM PMSF, 0.2 mM EDTA, and 25% glycerol) for 15 min on ice. The lysates were centrifugated at 13000×g for 10 min at 4°C. The supernatant containing the nuclear proteins was collected and frozen at −80°C until use. The protein content of nuclear fractions was determined with Bio-Rad protein assay. Synthetic double-strand oligonucleotides of consensus NF-κB binding sequence, 5′-AGTTGAGGGGACTTTCCCAGGC-3′ and 3′-TCAACTCCCCTGAAAGGGTCCG-5′, were 5′-end-labeled with biotin. Binding reactions containing 5 μg of nuclear proteins, double-distilled H2O, 2 μl 10-fold binding buffer, 2 μg poly (dI·dC) and 2 pmol oligonucleotide probe were incubated for 15 min at room temperature. Specific competition binding assays were performed by adding a 200-fold excess of an unlabeled probe as a specific competitor. Following formation of protein-DNA complexes, samples were loaded on a 6% nondenaturing polyacrylamide gel in 0.5× TBE buffer and then transferred to positively charged nitrocellulose membranes (Millipore, Bedford, MA, USA) by a transfer blotting apparatus and cross-linked in a Stratagene crosslinker. Gel shifts were visualized with streptavidin–horseradish peroxidase followed by chemiluminescent detection.
Transient transfection with Akt1 cDNA
Liposome-mediated transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) on AGS cells with a control pUSEamp empty vector (as the control group) or an expression construct for Akt1 cDNA in pUSEamp (activated) (Upstate Biotechnology, Lake Placid, NY, USA). Briefly, AGS cells (2 × 105 cells/well) were plated onto 6-well plates and transfected with the indicated plasmid at 60–70% confluence on the next day using Lipofectamine Plus reagent according to the manufacturer’s instructions. Briefly, lipofectamine (4 μl) and DNA (2 μg) were diluted in 100 μl of RPMI-1640 followed by equilibration at room temperature for 15 min after mixing. The lipofectamine–DNA complex was added to AGS cells, and being incubated for 12 h. Cells were then washed with PBS and replenished with RPMI-1640 containing 20% serum. Twelve hours after transfection, the cells were incubated with 2 μM nobiletin for 24 h. After a further 24 h of incubation, the media were removed, and cells were washed once with cold PBS. Finally, the transfected cells were collected and subjected to further studies.
Statistical analysis
Data were expressed as means ± standard deviation of three independent experiments and statistical analysis was obtained using a Student’s t-test. All statistical analyses of data were performed using Sigma Plot 2001 software (Systat Software Inc., San Jose, Calif., U.S.A.). Significant differences were established at P ≤ 0.05.
Results
Nobiletin inhibits the viability of HepG2, Caco-2, and AGS cells
In this study, we first examined the effect of nobiletin on cell viability in three cell lines, HepG2, Caco-2, and AGS cells. As shown in Fig. 1b, nobiletin showed a dose- and time-dependent inhibitory effect on the cell viability of three cancer cell lines. The strongest potency of nobiletin on the viability inhibitory effect of cancerous cells was toward AGS cells. Compared to the control group, after 24 and 48 h treatment with nobiletin at concentrations between 0 and 2 μM, the cell viability was not significantly altered, indicating nobiletin was not toxic to AGS cells at these dosages. When cells were treated with 2.5–4.5 μM nobiletin for 24 and 48 h, cell viability was significantly decreased. These results demonstrated treating with nobiletin with doses higher than 2 μM for 24 and 48 h resulted in dose- and time-dependent loss of cell viability in AGS cells, but doses lower than 2 μM for 24 and 48 h did not cause cytotoxicity. In contrast, the viable inhibitory effect of nobiletin on Chang liver cells was not significant when at the same concentrations. Next, we chose AGS cell line to perform for subsequent experiments.
The inhibition of cell adhesion, morphology/actin cytoskeleton arrangement, invasion, and migration by nobiletin
Previous studies have demonstrated that cancer metastasis and invasion are highly related to the degradation of ECM, intercellular adhesion, and cellular motility. We first examined the effect of nobiletin on cell adhesion of AGS cells by cell–matrix adhesion assay. Figure 2a showed that nobiletin exhibited a dose-dependent inhibitory effect on the cell adhesion ability of AGS cells. At 1.5 μM the cell adhesion was reduced to 78% and at 2 μM the cell adhesion was reduced to 61%. We next assessed the effect of nobiletin on the cell morphology and actin cytoskeleton arrangement by immunofluorescence staining assay. AGS cells were treated with different concentrations of nobiletin for 24 h. Actin fibers were stained with Alexa fluor 488-phalloidin (green, Excitation 495/Emission 519). As shown in Fig. 2b, when AGS cells were treated with 2 μM nobiletin, the cells exhibited changed morphology and became elongated, spindle shaped, and shrunken. Furthermore, to measure the effect of nobiletin on the invasive ability of AGS cells, a Boyden chamber coated with Matrigel was used in a dosage experiment. AGS cells treated with nobiletin (0, 1, 1.5, and 2 μM) for 48 h were plated on the upper chamber, and the number of cells which moved to the underside of the coated membrane were counted 8 h later under inverted microscopy. The result showed that the number of cells invaded to the lower chamber was significantly reduced by a 48 h treatment of nobiletin. Such reduction was concentration dependent with a 74% decrease (P < 0.005) when nobiletin was at 2 μM (Fig. 2c). The effect of nobiletin on AGS cell migration was further determined by wound-healing assay. As shown in Fig. 2d, an apparent and gradual increase of cells in the denude zone was observed in the cells treated with the control for 12, 24, 48 h under inverted microscope. According to a quantitative assessment, treatment with 1.5 and 2 μM of nobiletin inhibited 33 and 78% of cell migration after 24 h, respectively; and such doses of nobiletin inhibited 30 and 74% of cell migration at 48 h, respectively. Also, compared with the untreated cells, the level of AGS cells numbers decreased almost 4-fold when treating with 2 μM nobiletin for 48 h. The results demonstrated nobiletin significantly inhibited the invasion and migration of highly metastatic AGS cells.

The effects of nobiletin on the cell–matrix adhesion, cell morphology/actin cytoskeleton arrangement, invasion, and migration in AGS cells. Cells were treated with various concentrations (0, 1, 1.5, and 2 μM) of nobiletin and were then subjected to analyses for a cell–matrix adhesion, b immunofluorescence, c invasion, and d migration as described in “Materials and methods”. Values are expressed as mean ± SD of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 compared with the untreated control (dose 0); # P < 0.01, ## P < 0.001 compared with the 0 h-treated time
The inhibition of the expressions of MMP-2 and MMP-9 by nobiletin
The crucial role played by ECM degradation in cellular invasion suggests that matrix-degrading proteinases are required to clarify whether or not MMP2 and MMP-9 are involved in inhibition of invasion, motility, and adhesion by nobiletin. The effects of nobiletin on MMP-2 and MMP-9 activities were investigated by gelatin zymography under a condition of serum starvation, respectively. Figure 3a showed that, in the presence of 1, 1.5, and 2 μΜ of nobiletin, MMP-2 and MMP-9 activities were inhibited by nobiletin in a dose-dependent manner. The subsequent time course experiment showed that the reduction of MMP-2 and MMP-9 activities by nobiletin also occurred in a time-dependent manner when nobiletin was at 2 μΜ (Fig. 3b). The immunoblotting showed that the levels of MMP-2 and MMP-9 presented a progressive decrease when exposed to nobiletin treatment in a dose-dependent manner (Fig. 3c). In order to understand further the down-regulatory effects of nobiletin on proteases, RT-PCR was evaluated. It revealed that the treatment of 2 μΜ nobiletin for 24 h reduced significantly the mRNA levels of MMP-2 and MMP-9 (Fig. 3d). The nobiletin-mediated change in the mRNA levels MMP-2 and MMP-9 coincided with the protein levels as evidenced by western blot results, revealing that nobiletin might regulate MMP-2 and MMP-9 expressions at transcription levels. Furthermore, these results suggested that the antimetastatic effect of nobiletin was related to the inhibition of enzymatically degradative processes of tumor metastasis.

Effect of nobiletin on MMP-2 and MMP-9 expressions in AGS cells. a The activities of MMP-2 and MMP-9 were inhibited by nobiletin in a dose-dependent manner. b The activities of MMP-2 and MMP-9 were inhibited by nobiletin in a time-dependent manner. c The protein levels of MMP-2 and MMP-9 from whole-cell lysates were analyzed by western blot. β-Actin was used as loading control. d Cells were treated with various concentrations of nobiletin for 24 h. And then, RNA samples were extracted and subjected to a semi-quantitative RT-PCR for MMP-2 and MMP-9 with GADPH being an internal control
The inhibition of the phosphorylation of FAK/Akt and the protein expression of PI3K, small GTPases family by nobiletin
Several studies have indicated that FAK/PI3K/Akt is involved in the regulation of MMP-2 and MMP-9 activities on different cell types [16, 24]. To assess whether nobiletin mediates and/or inhibits the phosphorylation of FAK, Akt, the protein level of PI3K and the small GTPases family, AGS cells were treated with various concentrations of nobiletin for 6 h and 2 μM of nobiletin for various periods of time (0, 1, 3, and 6 h). Figure 4a and b showed nobiletin significantly inhibited the activation of FAK and Akt as shown by the decrease in the the phosphorylation of FAK and Akt. In addition, nobiletin inhibited the protein levels of PI3K, Ras, c-Raf, Rac-1, Cdc42, and RhoA in a dose- and time-dependent manner. On the other hand, the amount of RhoB protein gradually elevated with nobiletin treatment.

Dose- and time-dependent effect of nobiletin on the phosphorylation of FAK/Akt, the protein expressions of PI3K, and small GTPases family. In dose-dependent assay a, AGS cells were treated with various concentrations (0, 1, 1.5, and 2 μM) of nobiletin for 6 h. In time-dependent assay b, AGS cells were treated with 2 μM of nobiletin for 0, 1, 3, and 6 h. Activities of FAK phosphorylation, FAK, Akt phosphorylation, Akt, the expressions of PI3K, Ras, c-Raf, Rac-1, Cdc42, RhoA, and RhoB were analyzed by western blotting. β-Actin was used as a loading control
The inhibition of the DNA binding activity of NF-κB/IκBα degradation and phosphorylation by nobiletin
NF-κB of a transcriptional factor has been known to translocate to the nucleus and regulate the expressions of multiple genes involved in MMP-2/MMP-9 secretions. To clarify the involvement of NF-κB protein in the mechanism of nobiletin’s action, the effect of nobiletin on the DNA binding activity of NF-κB in AGS cells was investigated by EMSA. As shown in Fig. 5a, AGS cells were treated with 0–2 μM of nobiletin for 12 h, nobiletin inhibited NF-κB binding activity in a dose-dependent manner. Especially, the binding activity of NF-κB was inhibited by treatment with 2 μM of nobiletin. Furthermore, the expressions of NF-κB (p65 and p50) in the cytoplasmic and nuclear extracts were analyzed by western blotting to assess the possible inhibitory effect of nobiletin. As shown in Fig. 5b, nobiletin treatment resulted in a dose-dependent decrease in NF-κB (p65 and p50) protein level in the cytoplasmic as well as nuclear fraction. Result showed the cytoplasmic and nuclear level of NF-κB was gradually diminished at doses of 1, 1.5, and 2 μM nobiletin when compared to the 0 μM after treatment for 12 h. Also, the activation of NF-κB occurs through the phosphorylation of IκBα to release the NF-κB for nuclear translocation, and for binding to the promoter sites of target genes. To examine the effect of nobiletin on IκBα regulation, we investigated whether or not nobiletin had an effect on IκBα degradation and phosphorylation. As shown in Fig. 5c, nobiletin blocked the IκBα degradation by inhibiting phosphorylation of IκBα. Also, the intensity of western blotting reflected that nobiletin could enhance IκBα protein expression at a concentration >1 μM.

Effects of nobiletin on the NF-κB DNA binding activity/expression of NF-κB/IκBα phosphorylation and degradation in AGS cells. Cells were treated with various concentrations (0, 1, 1.5, and 2 μM) of nobiletin for 12 h, and then nuclear extracts were prepared and analyzed for a NF-κB DNA binding activity using biotin-labeled consensus NF-κB specific oligonucleotide. Then EMSA assay were performed as described in “Materials and methods”. Lane 1 nuclear extracts incubated with 100-fold excess unlabeled consensus oligonucleotide (comp.) to confirm the specificity of binding. Lanes 2–5 biotin-NF-κB plus various concentrations of nobiletin. Excess free probe is indicated at the bottom. Results from three repeated and separated experiments were similar. b Cytoplasmic and nuclear extracts were also analyzed by western blotting with anti-NF-κB (p65 and p50) antibodies. c Equal amounts of cytoplasmic proteins were resolved by SDS-PAGE, transferred to PVDF membrane, and probed with specific antibodies (anti-p-IκBα and anti-IκBα). β-Actin and C23 were used as loading controls. The densitometric results are expressed as mean ± SD of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 compared with the untreated control (dose 0)
The inhibition of the levels of phospho-Akt, MMP-2/9 and invasion/migration in Akt-transfected AGS cells by nobiletin
Our results have demonstrated that nobiletin could suppress cell invasion/migration, down-regulate FAK/PI3K/Akt signal, and reduce MMP-2/9 expression in AGS cells. To further investigate whether the inhibition of nobiletin mainly occurs through the inhibition of the FAK/PI3K/Akt signaling pathway, AGS cells were transiently transfected with active Akt. The western blotting results showed that the cells expressed as a control vector indeed diminished the levels of phospho-Akt and MMP-2/9 when cells were treated with nobiletin (Fig. 6a). The expression of constitutively active Akt also improved the abilities of invasion and migration in AGS cells which was originally inhibited by nobiletin as analyzed by Boyden chamber assay and wound-healing assay (Fig. 6b, c). After treatment with nobiletin, the Akt phosphorylation, MMP-2/9 expressions, and invasion/migration were decreased in the Akt-transfected AGS cells.

Effects of nobiletin inhibited the levels of phospho-Akt, MMP-2/9 and invasion/migration in Akt-transfected AGS cells. Cells were transfected with empty vector or Akt1 cDNA (activated) and treated with or without 2 μM of nobiletin for 24 h. a The cellular levels of phospho-Akt and MMP-2/9 were analyzed by western blot. β-Actin was used as an internal control. b Cell invasion and c migration were analyzed by Boyden chamber assay and wound-healing assay. The quantitative data were presented as means ± SD of three repeats from one independent study. * P < 0.05, ** P < 0.01 compared with the respective untreated group. (AGS/vector or AGS/Akt1 cDNA). ## P < 0.01 compared with invasive/migrated cells in nobiletin-treated AGS/vector group compared with nobiletin-treated AGS/Akt1 cDNA group
Discussion
Gastric cancer is a common malignancy in many countries of the world, especially in Asia. Prevention is likely to be the most effective means of not only reducing the incidence but also the mortality resulting from this disease. In recent years, research has been focused on the anti-cancer properties of pure components for application in chemotherapy. Chemoprevention is an active cancer preventive strategy to suppress, delay, or reverse human carcinogenesis. Nobiletin, the major constituent of Citrus genus, is implicated for its pharmacological property because of recent reports on its ability to inhibit cancer cell proliferation [25], induce cell-cycle arrest [26], and promote apoptosis [27]. The antitumor metastatic effect of nobiletin has been shown to be mediated by inhibiting the phosphorylation of MEK/ERK that is accompanied by a deceased expression level of MMP in HT-1080 cells [9]. In addition, nobiletin inhibited tumor promotion which was associated with inhibition of MAPK and Akt protein kinase cascades [5]. However, the molecular mechanism underlying the nobiletin-mediated inhibition of AGS cancer cell invasion and migration has not been elucidated until the present study. This study shows that the downregulation of FAK/PI3K/Akt and small GTPase signaling pathway might be responsible for the inhibitory effect of nobiletin.
During cancer progression, some tumor cells become motile and gain the capacity to attack the host tissue leading to metastasis. FAK can be activated in response to diverse stimuli and plays an important role in the proliferation and metastasis of cells [28]. Accumulated evidence suggests that FAK regulates focal adhesion signaling by phosphorylating multiple substrates and by acting as a scaffold for protein–protein interactions, as well as the matrix metalloproteinases (MMPs)-mediated matrix degradation, which in turn also regulates downstream signaling cascades [29–32]. Zeng et al. [33] reported that activated FAK induced PI3K is required for the production of matrix metalloproteinases (MMPs). Also, PI3K is one of the critical downstream signal molecules of FAK pathways [34]. Therefore, our results demonstrated that nobiletin inhibited the expressions of phospho-FAK, phospho-Akt, and PI3K. Shibata et al. [35] have shown that stimulated ovarian cancer cells with fibronectin activated MMP-9 secretion, while both antisense oligonucleotide to FAK and dominant-negative mutation of Ras abolished this phenomenon. On the other hand, Ras-homologous (Rho) GTPases play a pivotal role in the intracellular signaling network controlling differentiation, proliferation, cell survival, and metastasis. In human malignancies, Ras mutations are common, In fact, they have been identified in approximately 30% of cancers. Furthermore, the PI3K/Akt signaling pathway is also mediated by Ras [36]. In addition, the small GTPases family, including c-Raf, Rac-1, Cdc42, and RhoA, is critical in regulating the actin organization associated with cell motility, cell-cycle progression, gene expression, and apoptosis [14]. It is important to note that, RhoB belongs to the small GTPase family and supports a tumor-suppressive role. RhoB shares 86% amino acid sequence identify with RhoA, yet the roles of the low-molecular-weight GDP/GTP binding GTPases in oncogenesis are quite different. Past studies have evidenced that Ras suppressed RhoB expression in NH3T3 cells and human A549 lung cancer cells [37], and suppression of RhoB is a mechanism by which the Ras/PI3K/Akt pathway induces tumor survival, transformation, invasion, and metastasis [38]. In this study, the results showed decreased protein levels of Ras, c-Raf, Rac-1, Cdc42, and RhoA. However, the protein level of RhoB increased when AGS cells were treated with nobiletin. Based on the finding of decreased expression of Ras, c-Raf, Rac-1, Cdc42, and RhoA, further studies should be performed to evaluate the effects of nobiletin on the skeletal structure of the AGS cells. The results also showed a decrease in F-actin patterns when AGS cells were treated with nobiletin. All these findings indicated that nobiletin is involved in the inhibition of the downregulation of FAK/PI3K/Akt and small GTPase signals were observed after 6 h of incubation. Also, nobiletin reduced AGS cell adhesion, invasion, and migration at 24 or 48 h after incubation. Thus, our results suggested that the FAK/Akt/PI3K pathway and small GTPase family proteins were involved in nobiletin-induced suppression of invasion and migration. The present study has indicated that the PI3K/Akt signaling pathway induces the expression of NF-κB transcription factor [39]. Here, we have also found the treatment of nobiletin to AGS cells results in an inhibition of NF-κB DNA binding activity, accompanied by the inhibition of nuclear translocation of these factors. Thus, the inhibitory effect of nobiletin has a significant impact on the mechanism to inhibit the MMPs-mediated cellular events in AGS cells.
In summary, we proposed a schematic presentation of possible mechanism for the inhibitory effect of nobiletin on the invasion and migration of AGS cells. The Ras/PI3K/AKT signaling pathway is a possible mechanism of the inhibitory effects of nobiletin on AGS cells, including the increased protein level of cytoplasmic IκB which exerts inhibitory effects on the transcriptional factor NF-κB, subsequently decreasing MMP-2 and MMP-9 activities. Moreover, nobiletin can interfere with the rearrangement of the actin cytoskeleton by decreasing the expression of phospho-FAK, then resulting in antimetastatic effects (Fig. 7). The above findings and concepts disclosed here provide an important basis for a further exploration towards understanding the action mechanisms of nobiletin and possibly its beneficial effect in the prevention of tumor metastasis.

A proposed mechanism for the nobiletin suppresses invasion and migration of AGS cells (see the text for “Discussion”). Nobiletin could inhibit the activity of the FAK/PI3K/Akt pathway and decrease the expressions of Ras, c-Raf, Rac-1, Cdc 42, and Rho A, decreased the activation of NF-κB (p50 and p65) and increased protein level of cytoplasmic IκB, results in an inhibition of NF-κB DNA binding activity and MMP-2/MMP-9 gene expressions, and contributed to the inhibition of cell invasion and migration
Abbreviations
- MMPs:
-
Matrix metalloproteinases
- ECM:
-
Extracellular matrix
- PI3K/Akt:
-
Phosphoinositide 3-kinase/protein kinase B
- NF-κB:
-
Nuclear factor kappa B
- IκBα:
-
Inhibitor of kappaBα
References
Kelley JR, Duggan JM (2003) Gastric cancer epidemiology and risk factors. J Clin Epidemiol 56:1–9
Serafini M, Bellocco R, Wolk A, Ekstrom AM (2002) Total antioxidant potential of fruit and vegetables and risk of gastric cancer. Gastroenterology 123:985–991
Chen J, Montanari AM, Widmer WW (1997) Two new polymethoxylated flavones, a class of compounds with potential anticancer activity, isolated from cold pressed dancy tangerine peel in solids. J Agric Food Chem 45:364–368
Nagata U, Sakamoto K, Shiratsuchi H, Ishi T, Yano M, Ohta H (2006) Flavonoid composition of fruit tissues of citrus species. Biosci Biotechnol Biochem 70:178–192
Lai CS, Li S, Chai CY, Lo CY, Dushenkov S, Ho CT, Pan MH, Wang YJ (2008) Anti-inflammatory and antitumor promotional effects of a novel urinary metabolite, 3′,4′-didemethylnobiletin, derived from nobiletin. Carcinogenesis 29:2415–2424
Murakami A, Nakamura Y, Torikai K, Tanaka T, Koshiba T, Koshimizu K, Kuwahara S, Takahashi Y, Ogawa K, Yano M, Tokuda H, Nishino H, Mimaki Y, Sashida Y, Kitanaka S, Ohigashi H (2000) Inhibitory effect of citrus nobiletin on phorbol ester-induced skin inflammation oxidative stress, and tumor promotion in mice. Cancer Res 60:5059–5066
Nakajima A, Yamakuni T, Haraguchi M, Omae N, Song SY, Kato C, Nakagawasai O, Tadano T, Yokosuka A, Mimaki Y, Sashida Y, Ohizumi Y (2007) Nobiletin, a citrus flavonoid that improves memory impairment, rescues bulbectomy-induced cholinergic neurodegeneration in mice. J Pharmacol Sci 105:122–126
Kawabata K, Murakami A, Ohigashi H (2005) Nobiletin, a citrus flavonoid, down-regulates matrix metalloproteinase-7 (matrilysin) expression in HT-29 human colorectal cancer cells. Biosci Biotechnol Biochem 69:307–314
Miyata Y, Sato T, Imada K, Dobashi A, Yano M, Ito A (2008) A citrus polymethoxyflavonoid, nobiletin, is a novel MEK inhibitor that exhibits antitumor metastasis in human fibrosarcoma HT-1080 cells. Biochem Biophys Res Commun 366:168–173
Stupack DG (2007) The biology of integrins. Oncology 21:6–12
van Nimwegen MJ, van de Water B (2007) Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol 73:597–609
Mitra SK, Schlaepfer DD (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18:516–523
Bernhard EJ, Gruber SB, Muschel RJ (1994) Direct evidence linking expression of matrix metalloproteinase 9 (92-kDa gelatinase/collagenase) to the metastatic phenotype in transformed rat embryo cells. Proc Natl Acad Sci USA 91:4293–4297
Wennerberg K, Der CJ (2004) Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci 117:1301–1312
Hubchak SC, Sparks EE, Hayashida T, Schnaper HW (2009) Rac1 promotes TGF-beta-stimulated mesangial cell type I collagen expression through a PI3K/Akt-dependent mechanism. Am J Physiol Renal Physiol 297:F1316–F1323
Shih YW, Chen PS, Wu CH, Jeng YF, Wang CJ (2007) α-Chaconine-reduced metastasis involves a PI3K/Akt signaling pathway with downregulation of NF-κB in human lung adenocarcinoma A549 cells. J Agric Food Chem 55:11035–11043
Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-kappa B activity. Annu Rev Immunol 18:621–663
Ma Z, Shah RC, Chang MJ, Benveniste EN (2004) Coordination of cell signaling, chromatin remodeling, histone modifications, and regulator recruitment in human matrix metalloproteinase 9 gene transcription. Mol Cell Biol 24:5496–5509
Lu KW, Chen JC, Lai TY, Yang JS, Weng SW, Ma YS, Lu PJ, Weng JR, Chueh FS, Wood WG, Chung JG (2010) Gypenosides inhibits migration and invasion of human oral cancer SAS cells through the inhibition of matrix metalloproteinase-2/-9 and urokinase-plasminogen by ERK1/2 and NF-kappa B signaling pathways. Hum Exp Toxicol. doi:10.1177/0960327110372405
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Ochi Y, Atsumi S, Aoyagi T, Umezawa K (1993) Inhibition of tumor cell invasion in the Boyden chamber assay by a mannosidase inhibitor, mannostatin A. Anticancer Res 13:1421–1424
Hoppe-Seyler F, Butz K, Rittmuller C, von Knebel Doeberitz M (1991) A rapid microscale procedure for the simultaneous preparation of cytoplasmic RNA, nuclear DNA binding proteins and enzymatically active luciferase extracts. Nucleic Acids Res 19:5080
Ma W, Lim W, Gee K, Aucoin S, Nandan D, Kozlowski M, Diaz-Mitoma F, Kumar A (2001) The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide stimulated human macrophages. J Biol Chem 276:13664–13674
Chan KC, Ho HH, Huang CN, Lin MC, Chen HM, Wang CJ (2009) Mulberry leaf extract inhibits vascular smooth muscle cell migration involving a block of small GTPase and Akt/NF-kappaB signals. J Agric Food Chem 57:9147–9153
Luo G, Zeng Y, Zhu L, Zhang YX, Zhou LM (2009) Inhibition effect and its mechanism of nobiletin on proliferation of lung cancer cells. Sichuan Da Xue Xue Bao Yi Xue Ban 40:449–453
Ishii K, Tanaka S, Kagami K, Henmi K, Toyoda H, Kaise T, Hirano (2010) Effects of naturally occurring polymethyoxyflavonoids on cell growth, p-glycoprotein function, cell cycle, and apoptosis of daunorubicin-resistant T lymphoblastoid leukemia cells. Cancer Invest 28:220–229
Luo G, Guan X, Zhou L (2008) Apoptotic effect of citrus fruit extract nobiletin on lung cancer cell line A549 in vitro and in vivo. Cancer Biol Ther 7:966–973
Parsons JT (2003) Focal adhesion kinase: the first ten years. J Cell Sci 116:1409–1416
Canel M, Secades P, Garzo′n-Arango M, Allonca E, Suarez C, Serrels A, Frame M, Brunton V, Chiara MD (2008) Involvement of focal adhesion kinase in cellular invasion of head and neck squamous cell carcinomas via regulation of MMP-2 expression. Br J Cancer 98:1274–1284
Sein TT, Thant AA, Hiraiwa Y, Amin AR, Sohara Y, Liu Y, Matsuda S, Yamamoto T, Hamaguchi M (2000) A role for FAK in the Concanavalin A-dependent secretion of matrix metalloproteinase-2 and -9. Oncogene 19:5539–5542
Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia BP, Schlaepfer DD (2001) Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res 61:7079–7090
Mon NN, Ito S, Senga T, Hamaguchi M (2006) FAK signaling in neoplastic disorders: a linkage between inflammation and cancer. Ann NY Acad Sci 1086:199–212
Zeng ZZ, Jia Y, Hahn NJ, Markwart SM, Rockwood KF, Livant DL (2006) Role of focal adhesion kinase and phosphatidylinositol 3′-kinase in integrin fibronectin receptor-mediated, matrix metalloproteinase-1-dependent invasion by metastatic prostate cancer cells. Cancer Res 66:8091–8099
Choi YA, Lim HK, Kim JR, Lee CH, Kim YJ, Kang SS, Baek SH (2004) Group IB secretory phospholipase A2 promotes matrix metalloproteinase-2-mediated cell migration via the phosphatidylinositol 3-kinase and Akt pathway. J Biol Chem 279:36579–36585
Shibata K, Kikkawa F, Nawa A, Thant AA, Naruse K, Mizutani S, Hamaguchi M (1998) Both focal adhesion kinase and c-Ras are required for the enhanced matrix metalloproteinase 9 secretion by fibronectin in ovarian cancer cells. Cancer Res 58:900–903
Dancey JE (2002) Agents targeting Ras signaling pathway. Curr Pharm Des 8:2259–2267
Jiang K, Delarue FL, Sebti SM (2004) EGFR, ErbB2 and Ras but not Src suppress RhoB expression while ectopic expression of RhoB antagonizes oncogene-mediated transformation. Oncogene 23:1136–1145
Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S (2004) Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol Cell Biol 24:5565–5576
Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC, Tang CH (2009) CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochem Pharmacol 77:794–803
Acknowledgment
This work was supported by the grant from the Subsidized Project of the Chung Hwa University, Tainan, Taiwan (98-HT-08001).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Lee, YC., Cheng, TH., Lee, JS. et al. Nobiletin, a citrus flavonoid, suppresses invasion and migration involving FAK/PI3K/Akt and small GTPase signals in human gastric adenocarcinoma AGS cells. Mol Cell Biochem 347, 103–115 (2011). https://doi.org/10.1007/s11010-010-0618-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-010-0618-z