
Abstract
Toll-like receptor 4 (TLR4) activation has been implicated in the pathogenesis of myocardial ischemia/reperfusion (I/R) injury. The activated TLR4 is capable of activating a variety of proinflammatory mediators, such as tumor necrosis factor-a (TNF-a) and interleukin-6 (IL-6). Valsartan as a kind of Angiotensin II type 1 receptor blockers is gradually used for the treatment of ischemic heart disease depending on its anti-inflammation function. Therefore, we hypothesized that valsartan protects against myocardial I/R injury by suppressing TLR4 activation. We constructed the rat model of myocardial I/R injury. The rats were pretreated with valsartan for 2 weeks, and then subjected to 30 min ischemia and 2 h reperfusion. TLR4 and Nuclear factor kappa-B (NF-κB) levels were detected by quantitative real-time PCR and western blot. In order to evaluate myocardial damage, the myocardial infarct size, histopathologic changes, and the release of myocardial enzymes, proinflammation cytokines and Angiotensin II were analyzed by triphenyl tetrazolium chloride (TTC) staining, light microscopy, and enzyme-linked immunosorbent assay (ELISA), respectively. Valsartan preconditioning inhibited TLR4 and NF-κB expressions concomitant with an improvement in myocardial injury, such as smaller infarct size, fewer release of myocardial enzymes, and proinflammation mediators. These findings suggest that valsartan plays a pivotal role in the protective effects on myocardial I/R injury. This protection mechanism is possibly due to its anti-inflammation function via TLR4/NF-κB signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myocardial reperfusion therapy has become the optimal therapeutic strategy for ischemic heart disease, which can preserve myocardial viability and function by reversing myocardial ischemia and limiting the infarct size [1–3]. However, the subsequent ischemia/reperfusion (I/R) injury may reduce the therapeutic benefit [4]. Myocardial I/R is a complex pathophysiological process that involves various factors and pathways [5, 6]. Inflammatory response is considered to be a major cause of I/R-induced tissue injury [7]. There is comprehensive experimental and clinical evidence that anti-inflammatory actions attenuate I/R injury [8, 9].
Toll-like receptor 4 (TLR4) as a member of pattern recognition receptors plays an important role in the induction of inflammatory response by recognition of exogenous pathogen-associated molecular patterns and endogenous ligands [10, 11]. Activation of TLR4 is linked to expression of proinflammatory cytokines and activation of nuclear factor kappa B (NF-кB) signaling pathways in several cell types [10, 12, 13]. More recently, it was reported that both TLR4 knockout and TLR4 mutant mice exhibit less myocardial injury and inflammation after I/R compared to wild-type counterparts [14, 15]. In our previous study, we also found that TLR4 expression positively correlated with the levels of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in models of myocardial I/R rats [16].
Angiotensin II type 1 receptor blockers (ARBs) are extensively used for the treatment of cardiovascular disease as blood pressure-lowering agents [17–19]. Furthermore, more and more clinical and experimental studies have suggested that the cardioprotective effects of ARBs may extend to mechanisms beyond blood pressure lowering, such as anti-inflammation, anti-atherosclerosis, target organ protection, and so on [20–24]. Recently, some studies have demonstrated that ARBs treatment results in the inhibition of NF-κB activity and reduction of proinflammatory cytokines [25]. However, the anti-inflammation mechanism of ARBs and their effects on the proinflammation cytokine levels, TLR4 and NF-κB expressions, are still far from clear. In this study, we investigated the cardioprotective effects of Angiotensin II type 1 receptor blocker, valsartan, in rat myocardial I/R model to explore their anti-inflammation mechanism via TLR4/NF-κB signaling pathway.
Materials and methods
Animals
Male Sprague–Dawley (SD) rats (SPF class, 220–250 g) were purchased from Tongji Medical School, Huazhong University of Science and Technology (HUST), China. Animals were maintained under standard laboratory conditions at 25 ± 2°C, relative humidity of 50 ± 15%, and normal photoperiod (12 h dark and 12 h light). The procedures for experiments and animal care were approved by the Animal Care and Use Committee of HUST, and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (NIH Publication No. 80-23).
Drugs and preconditioning
Valsartan was kindly supplied by Beijing Novartis Pharma Ltd. (China), and dissolved in distilled water on the day of administration. The rats were randomly divided into four groups each of which contained 13 rats. Groups 1 and 2 were orally treated with vehicle (distilled water) for 2 weeks, which served as a sham control and an ischemia–reperfusion control. Groups 3 and 4 were orally administered with valsartan by gavage at dosages of 5 mg/kg daily (L-Val) and 10 mg/kg daily (H-Val) for 2 weeks before ischemia, respectively.
Myocardial I/R surgical procedure
Surgical ligation of the left anterior descending coronary artery (LAD) was performed similar to the methods described previously. Briefly, the animals were anesthetized with pentobarbital (30 mg/kg IP) [26] and, after tracheotomy, ventilation was provided using a breathing machine at a respiratory rate of 50/min with a tidal volume of 20 ml/kg body weight. Blood pressure was recorded from the left common carotid artery using a pressure transducer, and the heart rate was monitored by an electrocardiogram (ECG) during the procedure. A left parasternal incision was made through the third and fourth ribs, and the pericardium was gently opened to expose the heart. The LAD was ligated using a 6-0 silk suture. Additionally, a medical latex tube (socket, inner diameter, 1.5 mm) was placed between the ligature and the LAD. Myocardial ischemia was induced by compressing the LAD by tightening the ligature around the latex tube. The ECG was monitored for changes in the ST-T segment caused by tightening or loosing the ligature. After 30 min ischemia, the latex tube was removed to reperfuse the myocardium by restoring the coronary circulation. At 2 h post-reperfusion, rats were killed, and parts of the anterior wall of the left ventricular myocardium near the cardiac apex and blood samples were obtained for further analysis. The sham control group underwent the same procedures, with the exception of the induction of myocardial ischemia/reperfusion.
Biochemical studies
Blood serum samples were collected to measure the 3 specific marker enzymes, including the activities of creatine phosphokinase (CK), creatine phosphokinase-isoenzyme (CK-MB), and lactate dehydrogenase (LDH). All these marker enzymes were expressed as U/L using commercial kits (Beijing Kemeidongya Biotechnology Ltd., China).
Determination of infarct size
Five rats were sacrificed for the assessment of infarct area in each group. Two hours after the reperfusion period, rats' hearts were rapidly excised and sliced parallel to the atrioventricular groove into 2-mm-thick sections. The slices were incubated in 1% 2,3,5-triphenyl tetrazolium chloride (TTC) phosphate buffer (pH 7.4) for 20 min at 37°C. Infarct area in each heart determined by a computer-assisted image analysis system (Image-Pro Plus 3.0; Media Cybernetics, Silver Spring, MD) was multiplied by the thickness of the slice to calculate its volume. Infarct size was expressed as a percentage of left ventricular volume (%, infarct size/left ventricular).
Histologic examination
The formalin-fixed, paraffin-embedded sections of myocardial tissues were stained with hematoxylin and eosin and examined under a light microscope (≤400× magnification).
Quantitative real-time PCR analysis
The total RNA from the cardiac muscle samples was extracted and purified using the Trizol reagent kit (Invitrogen, US). Total RNA was reversely transcribed into complementary DNA (cDNA) using the cDNA synthesis kit (TaKaRa, Japan) according to the manufacturer’s protocol. The opticon-2 real-time PCR reactor (MJ Research, US) and real-time PCR kit (SYBR® Premix Ex TaqTM, TaKaRa, Japan) were employed based on the manufacturer’s instruction. The RT-PCR conditions were 42°C/15 min, 95°C/2 min for reverse transcription; polymerase chain reaction condition included pre-denaturing at 95°C for 10 s, then 40 cycles of 95°C for 5 s, and 60°C for 30 s and 72°C for 1 min. In this experiment, GAPDH and β-actin were used as the housekeeping genes. Levels of TLR4 mRNA and NF-κB mRNA were calculated based on the method of 2−ΔΔCt [27, 28]. The primers were as follows: TLR4, sense primer 5′-AGCCATTGCTGCCAACATCA-3′, antisense primer 5′-GCCAGAGCTACTCAGAAAC-3′. NF-κB, sense primer 5′-GGCAGCACTCCTTATCAA-3′, antisense primer 5′-GGTGTCGTCCCATCGTAG-3′. GAPDH, sense primer 5′-GACAACTTTGGCTCGTGGA-3′, antisense primer 5′-ATGCAGGGGTTCTGG-3′. β-actin, sense primer 5′-ACGTTGACATCCGTAAAGAC-3′, and antisense primer 5′-GAAGGTGGACAGTGAGGC-3′.
Western blotting
Cytoplasmic and nuclear protein extracts and membrane fractions were prepared from myocardial tissues as reported previously [29]. Western blotting was performed according to the manufacturer’s procedures. Briefly, 50 μg of cytoplasmic or nuclear proteins was separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. Non-specific binding sites were blocked with 5% non-fat dry milk in Tris-buffer saline (TBS)-0.05% Tween. The membrane was subsequently probed with primary antibody (anti-TLR4 or anti-NF-κB p65, diluted 1:200, Santa Cruz Biotechnology, USA) and incubated in horseradish peroxidase-conjugated secondary antibody (diluted 1:1000, Beijing Zhongshan Biotechnology, China). The protein bands were visualized by an enhanced chemiluminescence system [30], and β-actin was used as an internal control to correct the variations of different samples. The expression level of TLR4/NF-κB p65 was indicated as a ratio of TLR4/NF-κB p65 to β-actin.
Enzyme-linked immunosorbent assay
The titres of TNF-α, IL-6, and Angiotensin II in cardiac muscle samples were measured using enzyme-linked immunosorbent assay (ELISA), and the detailed manipulation process was performed according to the manufacturer's recommendations. The rat TNF-a and IL-6 ELISA kits were purchased from Shanghai Xitang Biotechnology Ltd., China. Cardiac levels of Angiotensin II were determined using commercial kit (Beijing North Biotechnology Ltd., China).
Statistical analysis
Quantitative data were expressed as mean ± SD. For experiments with three or more groups of animals, one-way analysis of variance (ANOVA) was performed, and Student–Newman–Keuls (SNK)-q test was used for selected groups. A P-value of less than 0.05 was considered as statistically significant. Analysis was carried out using Statistical Product and Service Solutions (SPSS) (Version 11.0).
Results
Effects of valsartan on serum marker enzymes
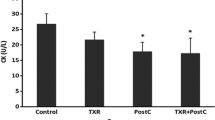
The activities of CK, CK-MB, and LDH in serum were used to monitor the damage of myocardium. Comparing with sham group, CK, CK-MB, and LDH activities in serum of control group markedly increased (P < 0.05). After preconditioning with valsartan at dosages of 5 and 10 mg/kg/day, the CK, CK-MB, and LDH levels in L-Val and H-Val groups decreased significantly compared with the control group (P < 0.05) (Table 1).
Hemodynamic parameters and infarct size assessment
There was no significant difference in heart rate and mean arterial pressure among the sham, control, L-Val, and H-Val groups (Fig. 1). Infarct size was found to be 37.8 ± 3.5% of the left ventricle in the control group. The infarct size was reduced to 23.7 ± 2.1% and 20.8 ± 2.9% of the left ventricle with treatment of valsartan at dosages of 5 and 10 mg/kg/day, respectively (P < 0.05) (Fig. 2). There was no significant difference between the L-Val and H-Val groups (P = 0.43, Fig. 2).

Hemodynamic parameters before and during myocardial I/R. a Heart rate change. b Mean arterial pressure change. T1 = before myocardial I/R, T2 = 30 min after myocardial ischemia, and T3 = at the end of myocardial I/R. Mean ± SD, n = 13

Myocardial infarct size in the four groups of rats. a Representative example of a heart slices from a rat stained with TTC. Infarct areas are not stained by TTC (white). b The infarct size at the end of reperfusion. S sham group, C control group, L-Val Low-dose valsartan group, and H-Val High-dose valsartan group. Mean ± SD, n = 5. △ P < 0.05, vs. S group; ★ P < 0.05, vs. C group
Light microscopic findings
The slides of histologic pathology demonstrated that the myocardium of healthy rats kept normal tissue structure and shape. Furthermore, the animals in the valsartan-treated groups had significantly less severe myocardium injury and inflammatory cells infiltration than did those in the control group (Fig. 3).

Histopathological detection of heart tissue changes in rats (magnification, ×400). a Sham group, the myocardial fibers are arranged in an orderly manner. b Control group, myocardial fibers are partially ruptured and lysed. c Low-dose valsartan group (L-Val), light edema is observed in the interstitial tissues. d High-dose valsartan group (H-Val), the histopathological changes are similar to L-Val
The role of valsartan in regulating expressions of TLR4 and NF-кB mRNA
The expression of TLR4 mRNA was significantly increased after myocardial I/R compared with sham group (3.97 × 10−2 ± 0.63 × 10−2 vs. 9.83 × 10−3 ± 1.28 × 10−3, P < 0.05). Treatment with valsartan 5 and 10 mg/kg/day could prevent the elevation of TLR4 mRNA level (2.67 × 10−2 ± 0.31 × 10−2 and 2.29 × 10−2 ± 0.40 × 10−2) after ischemia–reperfusion injury (Fig. 4). However, there was no difference between the groups L-Val and H-Val (P = 0.26). The expression of NF-κB mRNA paralleled that of TLR4 in the heart tissues (Fig. 4).

Expressions of TLR4 and NF-кB mRNA in the different experimental groups. S sham group, C control group; L-Val Low-dose valsartan group, and H-Val High-dose valsartan group. Mean ± SD, n = 8. △ P < 0.05, vs. S group; ★ P < 0.05, vs. C group
Effects of valsartan on TLR4 and NF-κB p65 proteins expression
Thirty minutes of ischemia and 2 h of reperfusion in control group significantly increased heart TLR4 and NF-κB p65 protein concentrations (1.36 ± 0.25 and 0.98 ± 0.17) compared with sham group (0.58 ± 0.10 and 0.32 ± 0.07, respectively) (P of both <0.05). Furthermore, in valsartan-treated animals, TLR4 (L-Val = 1.02 ± 0.11 and H-Val = 0.96 ± 0.14) and NF-κB p65 (L-Val = 0.73 ± 0.08 and H-Val = 0.60 ± 0.03) activities in tissues from the ischemic area was significantly lower compared with the control group (P both <0.05). However, no significant difference was found between valsartan groups (Fig. 5).

Effects of in vivo valsartan treatment on TLR4 and NF-κB p65 protein expressions after ischemia–reperfusion. a TLR4 protein expression. b NF-κB p65 protein expression. Original representative western blots are reported in the upper panels and relative levels of TLR4/NF-κB p65 in the lower panels. (1) heart in non-ischemic conditions; (2) heart subjected to ischemia–reperfusion; (3) heart subjected to ischemia–reperfusion from rats treated with 5 mg/kg/day valsartan; (4) heart subjected to ischemia–reperfusion from rats treated with 10 mg/kg/day valsartan. S sham group, C control group, L-Val Low-dose valsartan group, and H-Val High-dose valsartan group. Mean ± SD, n = 8. △ P < 0.05, vs. S group; ★ P < 0.05, vs. C group
Valsartan inhibited production of TNF-α and IL-6
The effects of valsartan on inflammatory cytokine expression in heart tissues of rats were analyzed by ELISA (TNF-α and IL-6). Comparing with sham group, Myocardial I/R induced a dramatic increase in the concentrations of TNF-α (41.13 ± 3.15 vs. 20.01 ± 1.87 pg/mg, P < 0.05) and IL-6 (57.66 ± 3.42 vs. 30.18 ± 2.09 pg/mg, P < 0.05) in the control group. Preconditioning with valsartan inhibited TNF-α (L-Val = 31.37 ± 1.46 pg/mg and H-Val = 28.36 ± 2.42 pg/mg) and IL-6 (L-Val = 42.60 ± 4.16 pg/mg and H-Val = 36.18 ± 1.75 pg/mg) production compared with control group (P both <0.05) (Fig. 6). In addition, the valsartan inhibitory effects of TNF-α and IL-6 were dose dependent (H-Val vs. L-Val, P both <0.05).

Effects of valsartan on TNF-a and IL-6 production in heart tissues. S sham group, C control group, L-Val Low-dose valsartan group, and H-Val High-dose valsartan group. △ P < 0.05, vs. S group; ★ P < 0.05, vs. C group; ▽ P < 0.05, vs. L-Val group. Values are presented as mean ± SD, n = 8
Angiotensin II levels in tissue
Angiotensin II concentration in control group was more than twice the concentration in the heart of the sham group rats (S = 46.76 ± 3.76 vs. C = 98.53 ± 7.47 pg/mg, P < 0.05). Comparing with the control group, only a tendency to increase the angiotensin II levels was observed in the valsartan groups (L-Val = 103.21 ± 10.73 pg/mg, H-Val = 111.86 ± 8.89 pg/mg, respectively).
Discussion
Clinically, reperfusion is the definitive treatment for acute coronary syndromes, especially for acute myocardial infarction. However, I/R injury which is characterized by a significant inflammatory response may lead to cardiac function recovery time delay. In this study, we showed that the pretreatment of valsartan before myocardial ischemia profoundly attenuated reperfusion injury by its action on inflammatory factors and related TLR4/NF-κB pathway in a rat myocardial I/R model. Myocardial enzyme release, infarct size, and morphological changes were significantly improved after reperfusion in valsartan-treated groups.
The most important finding in this study was that valsartan administration decreased TLR4 mRNA and protein expression, which were upregulated in the ischemia–reperfusion rats heart. The effect of TLR4 in myocardial I/R injury is supported by evidence of cardiovascular dysfunction in bacterial sepsis through TLR4 signaling [31, 32]. Under sterile conditions, however, noninfectious endogenous ligands released from injured cells or tissue fragments seem to initiate the inflammatory reaction by inducing TLR4 signaling [11, 33, 34]. Downstream of TLR4 signaling could occur through c-Jun N-terminal kinases (JNKs), p38 kinases, and NF-κB pathway. NF-κB is a key transcription factor in TLR4-mediated MyD88-dependent signaling pathway and plays a pivotal role in stimulating inflammatory cytokines, chemokines, and adhesion molecules secretion in myocardial I/R [35, 36]. In our study, we found the expression of NF-κB and proinflammatory cytokines (TNF-α, IL-6) was paralleled by TLR4 in the rat's heart, suggested that TLR4 plays an important role in recognizing endogenous ligands and triggering the inflammatory response in the process of myocardial I/R.
Valsartan, one of the most widely used renin–angiotensin system (RAS) inhibitors, along with its well-established actions on blood pressure, also exerts an anti-inflammatory effect [23]. As reported in hypertension patients, valsartan therapy could reduce the plasma level of serum inflammatory markers, such as IL-1 and high-sensitivity C reactive protein [37]. Interestingly, the results of our study revealed that valsartan administration could improve myocardial histopathological damage, reduce myocardial infarct size, and relieve inflammatory cytokines release by downregulating expression of TLR4/NF-κB mRNA and protein. Cytosolic enzymes leak into the intracellular space, where there is a cell membrane damage [38]. Therefore, enzyme analysis has proved considerably valuable in the diagnosis of myocardial infarction. Increased levels of CK, CK-MB, and LDH are well-known diagnostic markers of myocardial injury. In this study, marked elevations in the levels of these enzymes in the serum of the control group indicated that the occurrence of severe myocardial cell membrane damage compared with the sham group. Treatment with valsartan resulted in a significant reduction in the levels of these enzymes close to normal level compared with the control group, suggesting that valsartan can protect myocardial cells. Of course, the clinical setting where valsartan pretreatment might apply should be mentioned. Our results may be appropriate for those who develop an acute coronary syndrome while receiving ARBs for hypertension, heart failure, or diabetes as they might benefit from the cardioprotective effect of ARBs during reperfusion therapy.
This study demonstrated that myocardial I/R associated with a rise in cardiac Angiotensin II level. Moreover, such cardiac regulations were not prevented by Angiotensin II type 1 receptor blocker using valsartan. Our results were in agreement with previous data [39]. Several articles, however, reported opposite results. Silvestre et al. [40] reported that losartan prevented the myocardial infarction induced rise in tissue Angiotensin II level in rats. Therefore, the specific regulatory mechanism of the locally generated Angiotensin II is far from clear. We presumed that the elevated Angiotensin II in valsartan groups was mainly due to the effect of negative feedback after Angiotensin II type 1 receptor being blocked.
It is unclear why TNF-α and IL-6 concentration reduction exhibited a dose-dependent manner in valsartan-treated groups. In addition, we could find that TLR4/NF-κB mRNA and protein changes had no significant statistical difference between low-dose valsartan group and high-dose valsartan group. One possible explanation is that valsartan as Angiotensin II type 1 receptor blocker has another regulatory mechanism preventing inflammatory cytokines production. Recently, Dai and colleagues [41] reported that losartan could exert anti-inflammatory effects in spontaneously hypertensive rats by inhibition of PI3K/Akt. Furthermore, Angiotensin II receptor type I blockers are known to have direct anti-inflammatory actions. The direct effect is likely through their free radical scavenging properties, which are derived from their phenolic moiety [42]. Therefore, we presumed that protection by valsartan against myocardial I/R injury may be mediated partly by prevention of inflammatory mediators through the TLR4/NF-κB pathway.
In summary, our data contributed to a better understanding of the role of ARB in inhibiting the inflammatory response in the ischemia–reperfusion-induced myocardial injury. The increased production of TNF-α and IL-6 was associated with the elevated expression of TLR4/NF-κB in rat myocardial I/R model, and valsartan pretreatment could suppress their overexpression in heart tissues. Considering the important role in inflammation, a therapeutic approach involving the TLR4-mediated NF-κB signaling pathway might constitute a new strategy for myocardial ischemia–reperfusion injury.
References
Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI) (1986) Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet 1:397–402
ISIS-2 (Second International Study of Infarct Survival) Collaborative Group (1988) Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17, 187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 2:349–360
Cannon CP, Gibson CM, Lambrew CT, Shoultz DA, Levy D, French WJ et al (2000) Relationship of symptom-onset-to-balloon time and door-to-balloon time with mortality in patients undergoing angioplasty for acute myocardial infarction. JAMA 283:2941–2947. doi:10.1001/jama.283.22.2941
Ribichini F, Wijns W (2002) Acute myocardial infarction: reperfusion treatment. Heart 88:298–305. doi:10.1136/heart.88.3.298
Hansen PR (1995) Myocardial reperfusion injury: experimental evidence and clinical relevance. Eur Heart J 16:734–740
Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL (1998) Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 97:2259–2267
Frangogiannis NG, Smith CW, Entman ML (2002) The inflammatory response in myocardial infarction. Cardiovasc Res 53:31–47. doi:10.1016/S0008-6363(01)00434-5
Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B et al (2003) Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol 171:6856–6865
Serhan CN (2005) Novel omega-3-derived local mediators in anti-inflammation and resolution. Pharmacol Ther 105:7–21. doi:10.1016/j.pharmthera.2004.09.002
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature 388:394–397. doi:10.1038/41131
Miyake K (2007) Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin Immunol 19:3–10. doi:10.1016/j.smim.2006.12.002
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680. doi:10.1038/90609
Baumgarten G, Knuefermann P, Nozaki N, Sivasubramanian N, Mann DL, Vallejo JG (2001) In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis 183:1617–1624. doi:10.1086/320712
Oyama J, Blais C Jr, Liu X, Pu M, Kobzik L, Kelly RA et al (2004) Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 109:784–789. doi:10.1161/01.CIR.0000112575.66565.84
Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL et al (2004) Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg 128:170–179. doi:10.1016/j.jtcvs.2003.11.036
Yang J, Yang J, Ding JW, Chen LH, Wang YL, Li S et al (2008) Sequential expression of TLR4 and its effects on the myocardium of rats with myocardial ischemia-reperfusion injury. Inflammation 31:304–312. doi:10.1007/s10753-008-9079-x
Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U et al (2002) Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:995–1003. doi:10.1016/S0140-6736(02)08089-3
Brunner HR, Gavras H (2002) Angiotensin blockade for hypertension: a promise fulfilled. Lancet 359:990–992. doi:10.1016/S0140-6736(02)08062-5
Zannad F, Fay R (2007) Blood pressure-lowering efficacy of olmesartan relative to other angiotensin II receptor antagonists: an overview of randomized controlled studies. Fundam Clin Pharmacol 21:181–190. doi:10.1111/j.1472-8206.2007.00464.x
Navalkar S, Parthasarathy S, Santanam N, Khan BV (2001) Irbesartan, an angiotensin type 1 receptor inhibitor, regulates markers of inflammation in patients with premature atherosclerosis. J Am Coll Cardiol 37:440–444. doi:10.1016/S0735-1097(00)01138-4
Cianchetti S, Del Fiorentino A, Colognato R, Di Stefano R, Franzoni F, Pedrinelli R (2008) Anti-inflammatory and anti-oxidant properties of telmisartan in cultured human umbilical vein endothelial cells. Atherosclerosis 198:22–28. doi:10.1016/j.atherosclerosis.2007.09.013
Candido R, Allen TJ, Lassila M, Cao Z, Thallas V, Cooper ME et al (2004) Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109:1536–1542. doi:10.1161/01.CIR.0000124061.78478.94
Sironi L, Gelosa P, Guerrini U, Banfi C, Crippa V, Brioschi M et al (2004) Anti-inflammatory effects of AT1 receptor blockade provide end-organ protection in stroke-prone rats independently from blood pressure fall. J Pharmacol Exp Ther 311:989–995. doi:10.1124/jpet.104.072066
Varagic J, Frohlich ED, Susic D, Ahn J, Matavelli L, López B et al (2008) AT1 receptor antagonism attenuates target organ effects of salt excess in SHRs without affecting pressure. Am J Physiol Heart Circ Physiol 294:H853–H858. doi:10.1152/ajpheart.00737.2007
Chan YC, Leung PS (2007) Angiotensin II type 1 receptor-dependent nuclear factor-kappaB activation-mediated proinflammatory actions in a rat model of obstructive acute pancreatitis. J Pharmacol Exp Ther 323:10–18. doi:10.1124/jpet.107.124891
Maulik N, Engelman RM, Rousou JA, Flack JEIII, Deaton D, Das DK (1999) Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation 100:II369–II375
Marino JH, Cook P, Miller KS (2003) Accurate and statistically verified quantification of relative mRNA abundances using SYBR Green I and real-time RT-PCR. J Immunol Methods 283:291–306. doi:10.1016/S0022-1759(03)00103-0
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034
Altavilla D, Saitta A, Guarini S, Galeano M, Squadrito G, Cucinotta D et al (2001) Oxidative stress causes nuclear factor-kappaB activation in acute hypovolemic hemorrhagic shock. Free Radic Biol Med 30:1055–1066. doi:10.1016/S0891-5849(01)00492-0
Zheng YQ, Wei W (2005) Total glucosides of paeony suppresses adjuvant arthritis in rats and intervenes cytokine-signaling between different types of synoviocytes. Int Immunopharmacol 5:1560–1573. doi:10.1016/j.intimp.2005.03.010
Zeuke S, Ulmer AJ, Kusumoto S, Katus HA, Heine H (2002) TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc Res 56:126–134. doi:10.1016/S0008-6363(02)00512-6
Nemoto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA et al (2002) Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol 282:H2316–H2323
Beg AA (2002) Endogenous ligands of toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol 23:509–512. doi:10.1016/S1471-4906(02)02317-7
Dybdahl B, Wahba A, Lien E, Flo TH, Waage A, Qureshi N et al (2002) Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 105:685–690. doi:10.1161/hc0602.103617
Altavilla D, Deodato B, Campo GM, Arlotta M, Miano M, Squadrito G et al (2000) IRFI 042, a novel dual vitamin E-like antioxidant, inhibits activation of nuclear factor-kappaB and reduces the inflammatory response in myocardial ischemia-reperfusion injury. Cardiovasc Res 47:515–528. doi:10.1016/S0008-6363(00)00124-3
Shimamoto A, Chong AJ, Yada M, Shomura S, Takayama H, Fleisig AJ et al (2006) Inhibition of toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation 114:I270–I274. doi:10.1161/CIRCULATIONAHA.105.000901
Ridker PM, Danielson E, Rifai N, Glynn RJ, Val-MARC Investigators (2006) Valsartan, blood pressure reduction, and C-reactive protein: primary report of the Val-MARC trial. Hypertension 48:73–79. doi:10.1161/01.HYP.0000226046.58883.32
Mueller EA, Griffin WS, Wildenthal K (1977) Isoproterenol-induced cardiomyopathy: changes in cardiac enzymes and protection by methylprednisolone. J Mol Cell Cardiol 9:565–578. doi:10.1016/S0022-2828(77)80371-4
van Kats JP, Duncker DJ, Haitsma DB, Schuijt MP, Niebuur R, Stubenitsky R et al (2000) Angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade prevent cardiac remodeling in pigs after myocardial infarction: role of tissue angiotensin II. Circulation 102:1556–1563
Silvestre JS, Heymes C, Oubénaïssa A, Robert V, Aupetit-Faisant B, Carayon A et al (1999) Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 99:2694–2701
Dai Q, Xu M, Yao M, Sun B (2007) Angiotensin AT1 receptor antagonists exert anti-inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol 152:1042–1048. doi:10.1038/sj.bjp.0707454
Seeger H, Mueck AO, Lippert TH (2000) Effects of valsartan and 17 beta-estradiol on the oxidation of low-density lipoprotein in vitro. Coron Artery Dis 11:347–349. doi:10.1097/00019501-200006000-00008
Acknowledgment
We thank Jia-jun Wang from the Department of Immunology, Medical Science College of China Three Gorges University for his technical support and helpful suggestions on this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, J., Jiang, H., Yang, J. et al. Valsartan preconditioning protects against myocardial ischemia–reperfusion injury through TLR4/NF-κB signaling pathway. Mol Cell Biochem 330, 39–46 (2009). https://doi.org/10.1007/s11010-009-0098-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-009-0098-1