
Abstract
The aim of the current study was to determine the frequency of mutations in the beta-myosin heavy chain gene (MYH7) in a cohort of hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) and their families, and to investigate correlations between genotype and phenotype. About 130 consecutive patients diagnosed with HCM or DCM (69 with HCM and 61 with DCM) attending the cardiology clinic of Post Graduate Institute of Medical Education and Research were screened for mutations in the MYH7 gene. The control group for genetic studies consisted of 100 healthy subjects. We report 14 mutations in 6 probands (5 probands in HCM and 1 proband in DCM) and their family members. Out of these 6 mutations, 3 are new and are being reported for the first time. One known mutation (p.Gly716Arg) was found to be “de novo” which resulted in severe asymmetric septal hypertrophy (31 mm) and resulted in the sudden cardiac death (SCD) of the proband at the age of 21 years. Further, a DCM causing novel mutation p.Gly377Ser was identified which resulted in the milder phenotype. The present study shows that there is genetic and phenotypic heterogeneity of cardiomyopathies in Indian population. Further, the location and type of mutation in a given sarcomeric gene determines the severity and phenotypic plasticity in cardiomyopathies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cardiomyopathies are primary disorders of cardiac muscle associated with abnormalities of cardiac wall thickness, chamber size, contraction, relaxation, conduction, and rhythm. They are a major cause of morbidity and mortality at all ages and often result in heart failure. Over the past two decades, molecular genetic studies of humans and analyses of model organisms have made remarkable progress in defining the pathogenesis of cardiomyopathies. Hypertrophic cardiomyopathy has been found to result largely from mutations in sarcomere genes, whereas dilated cardiomyopathy is mainly caused by mutations in genes encoding contractile, cytoskeletal, and calcium regulatory proteins. Dissecting the complex genetic basis of different phenotypes of cardiomyopathy may be key to both better understanding and optimally managing these cardiovascular diseases. The discovery of causative mutations has important implications for diagnosis, prognosis, and intervention. India, with a population of more than a billion, has a large number of patients affected with cardiomyopathies; however, there is little information on spectrum of gene mutations and their association with disease phenotype and prognosis. In the present study, we have prospectively examined the spectrum of MYH7 mutations in 130 consecutive patients of HCM and DCM.
Material and methods
Patients
About 130 consecutive patients diagnosed with HCM and DCM (69 with HCM and 61 with DCM) attending the cardiology clinic of Post Graduate Institute of Medical Education and Research, were enrolled in the study after obtaining informed consent from all the participants. This study was approved by the Ethics committee of the institute. All the patients underwent physical examination, 12-lead ECG, and trans-thoracic two-dimensional echocardiography and Doppler studies.
About 100 healthy controls were also enrolled for the study.
Echocardiography
Standard views for M-mode and two-dimensional studies were obtained. For HCM patients end diastolic left ventricular wall thickness was recorded at the level of the mitral valve and papillary muscle in the anterior, posterior, septal, and lateral walls using short-axis two-dimensional images. Anterior and posterior septal thickness at the apex was assessed from apical short-axis view. The maximum left ventricular wall thickness was defined as the maximal measurement recorded in any of the myocardial segments studied. Left ventricle inflow and outflow velocities were determined using continuous and pulse wave doppler echocardiography.
Hypertrophic cardiomyopathy
Inclusion criteria
Individuals were diagnosed with HCM when echocardiography identified unexplained left ventricle hypertrophy greater than or equal to 13 mm or >2 standard deviations corrected for body surface area for children and adolescents.
Exclusion criteria
Patients with systemic hypertension and aortic stenosis or any other systemic or cardiac disease capable of producing the magnitude of wall thickening were excluded from the disease.
Dilated cardiomyopathy
Inclusion criteria
-
1.
Left ventricular ejection fraction (LVEF) ≤40% on echocardiography
-
2.
Absence of past history of myocardial infarction or coronary artery disease.
-
3.
Absence of secondary cause of left ventricular dysfunction including primary valvular heart disease, ventricular outflow tract obstruction, and coronary artery anomalies.
-
4.
Coronary angiography was carried out in all patients more than 35 years of age and in young individuals if clinically indicated.
Exclusion criteria
Patients with concomitant disease like infection, autoimmune disease, cancer, as well as patients with coronary artery disease (CAD) and with advanced chronic renal failure (CRF) were excluded.
Amplification of genomic DNA
The DNA was prepared from peripheral blood leukocytes by proteinase-phenol -chloroform extraction method. Primers for amplification reactions were designed by Primer3 software. Exons 8–24 of MYH7 gene (Accession no. M57965.2) were amplified using intronic primers flanking these exons according to the reported sequences of the MYH7. Primer sequences and PCR amplification conditions are available on request.
Denaturing high performance liquid chromatography (DHPLC)
After PCR, the amplified samples were directly analyzed by DHPLC (Transgenomic WAVE system Model 4500) using UV detection. Eight microliters of the PCR product was injected to reverse-phase DNASepR cartridges (Transgenomic USA). PCR products were eluted from the column by a linear acetonitrile gradient in 0.1 mM TEAA buffer (Transgenomic, USA), pH 7 at a constant flow rate of 0.9 ml/min using Rapid DNA programme. Run time was 4 min per sample. These conditions were used based on WAVEMAKER software (Supplementary online materials).
Sequencing
Variants identified by DHPLC were amplified again and PCR products were purified prior to sequencing using a kit from Amersham Pharmacia Biotech. Sequencing was performed using a Big Dye Terminator Cycle Sequencing Kit from Applied Biosystems Inc. The sequences were analyzed on an ABI Prism 3100 Sequencer in accordance with the manual of the manufacturer. The identified mutation was reconfirmed by taking a second blood sample from the affected patient and reanalyzing the amplified PCR product by sequencing (Supplementary online materials).
Statistical analysis
All statistical analyses were performed using SPSS (Statistical Package for Social Sciences) for windows (Version 10). Discrete and continuous variables were compared between patients and controls using Pearson’s χ2 test and unpaired t-test as appropriate. P values were subject to Bonferroni’s correction and considered significant when <0.05.
Results
Clinical characteristics
The clinical characteristics of the patients are given in Table 1. At presentation 25.86% of the patients were in New York Heart Association (NYHA) class III and IV. While 67.24% patients had dyspnea, 56.89% had angina pectoris and 25.28% of the patients had at least one event of syncope. ECG was abnormal in 64% of HCM patients and 72% of DCM patients. The mean septal thickness was 22.68 ± 5.17 mm for HCM patients. The mean LVEF at enrollment was 37.57 ± 12.09% for DCM patients.
Mutations
We identified 6 MYH7 mutations (5 in HCM and 1 in DCM). Table 2 shows the location, type, and associated phenotype of these mutations. Out of these 6 mutations, 3 are new and are being reported for the first time. One known mutation was found to be “de novo.”
Genotype phenotype correlations
p.Tyr266Cys
This mutation was observed in Proband H47. Echocardiography of the proband (Fig. 1) revealed severe left ventricular hypertrophy with septal thickness of 32 mm and involvement of right ventricle. This mutation has been identified for the first time in an HCM proband. The mutation lies in the conserved N-terminal region of MYH7 and results in substitution of tyrosine with cysteine. This mutation lies on the junction of exon 9 and 10, so it might be affecting the splicing of the pre mRNA of MYH7 which results in expression of aberrant MYH7 protein. The contractility of heart depends upon various sarcomeric proteins which work in a harmonious way. A mutant MYH7 leads to disruption of this harmony and hence results in a hypertrophied response. This was also evident from the observed phenotype of the proband. She had severe left ventricular hypertrophy with the involvement of right ventricle, indicative of a severe phenotype. Bioinformatic analysis showed that this mutation lies in the upper 50 kDa domain which is involved in the actin binding. The proper interaction of actin and myosin is very important for the ATPase activity of the myosin and for pulling of actin thin filaments by myosin. Any disruption of this interaction results in abnormal pulling of thin filaments over thick filaments and hence abnormal contractility. This leads to hypertrophic response as was seen in the case of present proband with this mutation.

Echocardiography of the proband H47 showing severe left ventricular hypertrophy
We could not confirm if this mutation was familial or “de novo” as other family members of the proband were not available for genotyping.
p.Gly377Ser
This mutation was seen in Proband D89. Echocardiography of the proband revealed dilated ventricles. Family history revealed sudden cardiac death (SCD) of the father of the proband who died suddenly at the age of 50 years. The mutation lies in the conserved N terminal region of MYH7 and results in substitution of glycine with serine. Five other family members aged 38, 28, 20, 15, and 12 years were found to be carriers of this novel mutation and were phenotypically normal (Fig. 2). Hence, the mutation was classified as familial. The LVEF of the patient has improved from 32% at presentation to 52% presently. Since several young members were identified as carriers and they are disease free, the mutation appears to be associated with a variable penetrance and favorable disease course. The absence of disease in the mutation carriers shows that other environmental factors and disease modifiers are involved in the pathogenesis of the dilated cardiomyopathy. The absence of disease in the mutation carriers shows that other environmental factors and disease modifiers are involved in the pathogenesis of the dilated cardiomyopathy.

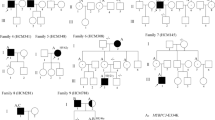
Pedigree of Proband D 89: only generations III and IV were available for genotyping, Filled symbol with arrow is proband, partially filled symbols are asymptomatic carriers, slashed is deceased individuals, ? = status unknown
p.Met515Thr
This mutation was identified in proband H10. Echocardiography of the proband with p.Met515Thr mutation revealed that she had asymmetrical septal hypertrophy with septal thickness of 19 mm. Echocardiography video of this proband was not available. She died at the age of 20 due to sudden cardiac death. The mutation lies in the relay domain of MYH7. During the recovery stroke, the myosin motor is primed for the next power stroke by a 60° rotation of its lever arm [1]. This reversible motion is coupled to the activation of the ATPase function of myosin through conformational changes along the relay helix, which runs from the Switch-2 loop near the ATP to the converter domain carrying the lever arm. The flexibility changes in the converter and relay domains result in the anomalous ATPase function of MYH7. The mutation appears to be associated with high risk of sudden cardiac death. A previously described different mutation Met515Arginine has also been reported at the same position by Van Driest et al. [2]. The phenotype was not described in the reference. The family of the proband was not available for the study.
p.Val606Met
This mutation was identified in proband H74. Echocardiography of the proband (Fig. 3) revealed concentric left ventricular hypertrophy with septal thickness of 24 mm. Pedigree of the family is shown in Fig. 4. This is a known mutation. It results in substitution of valine with methionine. It lies in the upper 50 kDa domain. Both the children of the proband were found to be carriers of the mutation. Hence, the mutation was classified as familial. On evaluation, the son of the proband, aged 25 years was found to have breathlessness on exertion and climbing two floors. Echocardiography of the son (Fig. 5) showed that he had HCM with maximal septal thickness of 13 mm. The daughter of the proband aged 19 years was found to be asymptomatic with maximal septal thickness of 9 mm.

Echocardiography of the proband showing concentric left ventricular hypertrophy

Filled symbol with arrow is proband, filled symbol is symptomatic carrier, partially filled symbols are asymptomatic carriers, slashed is deceased individuals, ? = status unknown

Echocardiography of the proband’s son
There have been conflicting reports regarding phenotypic association of this mutation. A few studies have observed this mutation to be associated with mild phenotype and as the prototype of a “benign” MYH7 mutation [3]. However, Fannapazir et al. [4] reported this mutation to be associated with severe phenotype: a family with four out of eight affected carrier members died of SCD. Nakajima et al. [5] also reported several premature deaths in persons with this mutation. So there, is large variability of phenotypes associated with this mutation. This may be due to the effect of modifier genes and environmental factors. Based on the previous published literature and our results, the mutation appears to be associated with variable phenotypic expression of the disease.
p.Gly716Arg
This mutation was identified in proband H91. Echocardiography of proband (Fig. 6) revealed severe asymmetrical septal hypertrophy with septal thickness of 31 mm. Pedigree of the family is shown in Fig. 7. Both father and mother of the proband were not found to carry this mutation; hence, the mutation was classified as “de novo.” Paternity was confirmed by STR analysis of 15 microsatellite markers. The proband died due to SCD at the age of 21 years.

Echocardiography of the proband showing severe asymmetrical septal hypertrophy

Pedigree of Family H91 with p.Gly716Arg mutation. Filled symbol with arrow is proband. SCD is sudden cardiac death
This is the first time that this mutation has been found to be “de novo.” This suggests that mutations in the sarcomeric genes can be familial as well as “de novo” and certain regions in these genes are more prone to mutations and occur as “hot spots.” There are consistent reports suggesting phenotypic associations of this mutation with severe phenotype. Hwang et al. [6] reported four SCD in 13 affected individuals in a family. Van Driest et al. [2] also reported sudden cardiac death at a young age in a patient with this mutation. Based on the previous published literature and our results, the mutation appears to be associated with severe asymmetrical hypertrophic cardiomyopathy and high risk of SCD. The mutation lies in the converter domain of MYH7. The rotation of the converter domain is essential for ATP hydrolysis. The flexibility changes in the converter domain results in the anomalous ATPase function of MYH7 and is expected to lead to a severe phenotype. The mechanisms through which this mutation leads to severe phenotype remains to be examined in vivo.
p.Arg787His
This mutation was identified in proband H61. The proband was diagnosed during routine examination for abnormal ECG. This is a known mutation [7]. It results in substitution of arginine with histidine. It lies in the neck domain of MYH7. The phenotype of the mutation carrier was not described in the previous report [7]. Echocardiography of the proband (Fig. 8) revealed apical hypertrophy with a maximum thickness of 18 mm. The mutation appears to be associated with favorable disease course. The proband did not participate in further screening process. Only sample A of the proband was tested. Because of the non-availability of the family of the proband for genetic analysis, we could not classify this mutation as familial or “de novo.”

Echocardiography of the proband showing apical hypertrophy
Discussion
Dissecting the complex genetic basis of cardiomyopathy is important for both better understanding and optimally managing these genetic cardiovascular diseases. Very little data are available on spectrum of mutations, genotype–phenotype correlations, and prognosis in Indian patients with mutations in MYH7 gene. Till date, only two MYH7 mutations, homozygous R870H and MYH7 DeltaE927, have been reported from HCM patients from South India [8, 9].
We observed allelic heterogeneity of MYH7 mutations; 2 MYH7 mutations at different locations resulted in two different phenotypes: a mutation at position 515 (p.Met515Thr) led to HCM and a mutation at 377 (p.Gly377Ser) caused DCM. These observations confirm genetic overlap in different idiopathic cardiomyopathies and indicate that different mutations in the same gene could lead to different cardiomyopathy phenotypes. The final phenotype may ultimately depend on the (i) location of the mutation in a given protein and (ii) the type of amino acid substitution. For example, mutation in an arginine residue (Arg145) located in the inhibitory domain of TNNI3 has been found to result in either HCM or RCM depending on the amino acid substitution: Arg145Gly and Arg145Gln have been found to be associated with HCM [10, 11], whereas Arg145Trp is associated with RCM [12].
The published literature shows a wide spectrum of heterogeneity in mutations for different populations; this necessitates the identification of population specific mutations. In the present study, we prospectively investigated 130 cardiomyopathy patients for MYH7 mutations and the influence of these mutations on phenotype and prognosis of the disease. We identified six mutations: three novel and three known mutations in MYH7 in our cohort. The identification of these new mutations suggests that not all known mutations associated with cardiomyopathy have been detected and there is need for identification of population specific mutations in different ethnic populations.
These mutations represent dominant negatives by disturbing contractile function despite the production of a normal protein by the remaining normal allele. Consistent with this conclusion is the finding that mutant beta-myosin separated from the heart muscle in cases of hypertrophic cardiomyopathy translocate actin filaments with an abnormally low sliding velocity in motility assays in vitro [13] possibly due to ethnic variations and environmental factors.
We found mutations in MYH7 in both cardiomyopathy phenotypes, i.e., in HCM and DCM. The association of these mutations with phenotypes ranged from asymmetrical septal hypertrophy to concentric and apical hypertrophy and with both benign and severe forms of the disease, indicating that there is a wide genetic and phenotypic heterogeneity of HCM and DCM in our population similar to that reported for other ethnic populations such as Caucasians and Japanese [3, 5, 11, 14–16].
Traditionally, it has been assumed that HCM and DCM develop during adolescence, to reach full morphologic disease expression at the time of physical maturity (≈18 years). The present study, however, suggests that the disease progression may be more complex. The onset of disease varied from second to fifth decade of life in patients with MYH7 mutations. For example, an HCM proband (H91) had a very early onset of symptoms in the second decade and a proband (H61) was diagnosed with apical hypertrophy in the fifth decade of his life. These observations indicate heterogeneity in genotype and course of the disease.
It has been reported that not all the individuals with a mutation in one of the cardiomyopathy genes will express the clinical features during their lifetime. Our data also indicate that there is considerable variation in penetrance between different mutations within the same gene and even between close relatives in the same family carrying the same specific mutation. With increasing knowledge in molecular genetics and increasing numbers of reported families with different genetic defects, the estimated figures of penetrance might very well change in the future. For the majority of disease causing mutations, the reported numbers of patients are too low to be able to give accurate estimates of penetrance.
The prevalence of MYH7 mutations were found to be lower in our cohort (HCM 7.2% and 1.6% in DCM) as compared to those observed in other populations [3, 5, 11, 14–16]. This difference may be due the fact that MYH7 gene mutations may not be predominant in Indians and some other sarcomeric and yet to be identified genes may be involved in the pathophysiology of cardiomyopathy. Our results also suggest that each population may have its own specific mutations.
Conclusion
The present study shows that there is genetic and phenotypic heterogeneity of cardiomyopathies in Indian population. Further, the location and type of mutation in a given sarcomeric gene determines the severity and phenotypic plasticity in cardiomyopathies.
References
Koppole S, Smith JC, Fischer S (2006) Simulations of the myosin II motor reveal a nucleotide-state sensing element that controls the recovery stroke. J Mol Biol 361:604–616. doi:10.1016/j.jmb.2006.06.022
Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004) Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:602–610. doi:10.1016/j.jacc.2004.04.039
Havndrup O, Bundgaard H, Andersen PS, Larsen LA, Vuust J, Kjeldsen K, Christiansen M (2001) The Val606Met mutation in the cardiac beta-myosin heavy chain gene in patients with familial hypertrophic cardiomyopathy is associated with a high risk of sudden death at young age. Am J Cardiol 87:1315–1317. doi:10.1016/S0002-9149(01)01532-6
Fananapazir L, Epstein ND (1994) Genotype–phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation 89:22–32
Nakajima-Taniguchi C, Azuma J, Nagata S, Kishimoto T, Yamauchi-Takihara K (1995) A missense mutation in the beta-myosin heavy chain gene in a Japanese patient with hypertrophic cardiomyopathy. Jpn Circ J 59:833–837
Hwang TH, Lee WH, Kimura A, Satoh M, Nakamura T, Kim MK, Choi SK, Park JE (1998) Early expression of a malignant phenotype of familial hypertrophic cardiomyopathy associated with a Gly716Arg myosin heavy chain mutation in a Korean family. Am J Cardiol 82:1509–1513. doi:10.1016/S0002-9149(98)00695-X
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M (2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107:2227–2232. doi:10.1161/01.CIR.0000066323.15244.54
Tanjore RR, Sikindlapuram AD, Calambur N, Thakkar B, Kerkar PG, Nallari P (2006) Genotype–phenotype correlation of R870H mutation in hypertrophic cardiomyopathy. Clin Genet 69:434–436. doi:10.1111/j.1399-0004.2006.00599.x
Bashyam MD, Savithri GR, Gopikrishna M, Narasimhan C (2007) A p.R870H mutation in the beta-cardiac myosin heavy chain 7 gene causes familial hypertrophic cardiomyopathy in several members of an Indian family. Can J Cardiol 23:788–790
Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T (1997) Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 16:379–382. doi:10.1038/ng0897-379
Mogensen J, Murphy RT, Kubo T, Bahl A, Moon JC, Klausen IC, Elliott PM, McKenna WJ (2004) Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:2315–2325. doi:10.1016/j.jacc.2004.05.088
Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, Gimeno JR, Elliott P, McKenna WJ (2003) Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 111:209–216
Cuda G, Fananapazir L, Zhu WS, Sellers JR, Epstein ND (1993) Skeletal muscle expression and abnormal function of beta-myosin in hypertrophic cardiomyopathy. J Clin Invest 91:2861–2865. doi:10.1172/JCI116530
Liu SX, Hu SJ, Sun J, Wang J, Wang XT, Jiang Y, Cai J (2005) Characteristics of the beta myosin heavy chain gene Ala26Val mutation in a Chinese family with hypertrophic cardiomyopathy. Eur J Intern Med 16:328–333. doi:10.1016/j.ejim.2005.02.008
Nishi H, Kimura A, Harada H, Koga Y, Adachi K, Matsuyama K, Koyanagi T, Yasunaga S, Imaizumi T, Toshima H (1995) A myosin missense mutation, not a null allele, causes familial hypertrophic cardiomyopathy. Circulation 91:2911–2915
Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, Tajik AJ, Gersh BJ (2002) Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation 106:3085–3090. doi:10.1161/01.CIR.0000042675.59901.14
Acknowledgments
TSR, SA and TSA received SRF from ICMR, India.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Rai, T.S., Ahmad, S., Bahl, A. et al. Genotype phenotype correlations of cardiac beta-myosin heavy chain mutations in Indian patients with hypertrophic and dilated cardiomyopathy. Mol Cell Biochem 321, 189–196 (2009). https://doi.org/10.1007/s11010-008-9932-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9932-0