
Abstract
Cardiac hypertrophy is an independent risk factor in the development of heart failure. However, the cellular mechanisms underlying the transition from compensated hypertrophy to heart failure are incompletely understood. The aim of this study was to investigate changes in myocardial substrate utilisation and function in pressure-overload hypertrophy (using 13C NMR spectroscopy) in parallel with alterations in the expression pattern of genes involved in cardiac fatty acid and glucose uptake and oxidation. Left ventricular hypertrophy was induced surgically in Sprague–Dawley rats by inter-renal aortic constriction. Nine weeks later, hearts were perfused in the isovolumic mode with a physiological mixture of substrates including 5 mM 1-13C glucose, 1 mM 3-13C lactate, 0.1 mM U-13C pyruvate and 0.3 mM U-13C palmitate and cardiac function monitored simultaneously. Real-time PCR was used to determine mRNA levels of PPARα and PPARα-regulated metabolic enzymes. Results showed that at the stage of compensated hypertrophy, fatty acid oxidation (FAO) and expression of genes involved in FAO were markedly reduced, whilst pyruvate oxidation was enhanced, highlighting the fact that metabolic remodelling is an early event in the development of cardiac hypertrophy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Left ventricular hypertrophy is the adaptive response of the heart to chronic mechanical overload. Although initially beneficial, the long-term effects of ventricular hypertrophy lead to functional deterioration and heart failure [1]. Indeed, cardiac hypertrophy is an independent risk factor in the development of heart failure [2] and a major cause of morbidity and mortality worldwide [3]. The prevalence of hypertrophic phenotype (due to pre-existing hypertension) in patients with heart failure is an alarming 69% in the UK [4] and 74% in the US [5]. Despite the enormity of this growing problem, the cellular mechanisms underlying the transition from compensated hypertrophy to heart failure are incompletely understood.
Adaptations in cardiac energy metabolism [1, 6] are considered a key event in the hypertrophic remodelling process, as are changes in extracellular matrix [7], myocardial function [8] and calcium handling proteins [9]. In particular, cardiac hypertrophy is characterised by a switch in the substrate preference from fatty acids (FA) towards carbohydrates [10–12]. As energy production is tightly coupled to substrate oxidation [1], sustained changes in the profile of myocardial substrate selection may have deleterious functional consequences in the hypertrophied myocardium. Only recently, have experimental and clinical studies highlighted the potential role of metabolic remodelling in the progression of hypertrophy to failure [6, 13]. However, many studies to date have investigated substrate oxidation in the hypertrophied myocardium using primarily glucose, palmitate and lactate as substrates, often excluding pyruvate—a significant myocardial fuel [14, 15]. Thus, the profile of substrate oxidation in the compensated hypertrophied heart using a more physiological mixture of substrates has yet to be determined.
Previous studies have shown that end-stage heart failure is characterised by a down-regulation of mRNA and protein expression of FAO enzymes [16–18]. However, alterations in the profile of metabolic gene expression are less clearly defined in the compensated hypertrophied heart. Consequently, dissecting the transcriptional mechanisms governing energy providing pathways in the adaptive phase of cardiac hypertrophy is vital to decipher the evolution of heart failure.
The aim of this study, therefore, was to investigate adaptations in energy substrate utilisation (using 13C NMR spectroscopy) and myocardial function in the compensatory phase of hypertrophy in parallel with alterations in the expression pattern of genes involved in cardiac FA and glucose uptake and oxidation. Our findings demonstrate significant metabolic remodelling in the adaptive phase of cardiac hypertrophy characterised by a marked decline in FAO with a concomitant down-regulation of FAO genes. This is paralleled by a compensatory increase in pyruvate oxidation and maintenance of cardiac function, indicating that, in the short-term, these adaptations are beneficial to the hypertrophied heart.
Materials and methods
Aortic constriction
All animal experimentation conformed to the UK Animals (Scientific Procedures) Act 1986. Left ventricular hypertrophy was induced surgically in male Sprague–Dawley rats (220–250 g, Charles River Inc. Kent), by abdominal aortic constriction as described previously [19] following anaesthesia with isoflurane in oxygen (3% in 3 l/min). A 0.5 mm outer diameter blunted needle was used to constrict the abdominal aorta between the left and right renal arteries. Control animals underwent the same procedure but without constriction of the aorta. Animals were maintained for 9 weeks post surgery in a 12:12 h light–dark cycle and provided with food and water ad libitum. The degree of hypertrophy was evaluated by determining the heart weight to tibia length ratio [20].
Isolated heart preparation
Following intra-peritoneal anaesthesia with sodium thiopentone (125 mg/kg body weight), hearts were rapidly excised and perfused in a modified isovolumic Langendorff mode with Krebs–Henseleit bicarbonate buffer equilibrated with 95% O2–5% CO2 (37°C, pH 7.4) [21], at a constant coronary flow rate (14 ml/min). Buffer contained 3% bovine serum albumin (essentially fatty acid free, Serologicals, USA), and the following components (mmol/l): NaCl 118, KCl 4.8, NaHCO3 25, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.25, glucose 5, sodium lactate 1, sodium pyruvate 0.1, sodium palmitate 0.3, glutamine 0.5 and insulin (0.1 mU/ml). Cardiac function was monitored simultaneously via a fluid-filled balloon inserted into the left ventricle and connected to a physiological pressure transducer (SensoNor 840) and a 2-Channel MacLab/2e system (AD Instruments, Hastings, UK). End-diastolic pressure was set to approximately 5 mmHg by adjusting the balloon volume. Oxygen content (mmHg) of the influent and effluent perfusates from the oxygenator and the pulmonary artery, respectively, was measured at 15-min intervals using a blood–gas analyser (ABL77 Radiometer, Copenhagen, Denmark) and myocardial oxygen consumption [MVO2 (μmole/min/g wet weight)] calculated as the product of arterio-venous oxygen content difference and coronary flow rate (ml/min) normalised to wet heart weight [22]. Left ventricular developed pressure [LVDP (mmHg)] and heart rate [HR (beats/min)] were recorded and the rate pressure product [RPP = LVDP × HR (mmHg/min)] calculated. The ratio of RPP/MVO2 was used as an indicator of cardiac efficiency.
Experimental protocol
After a 20 min equilibration, the perfusion medium was switched to an identical buffer (in terms of components and their concentrations) but containing palmitate labelled uniformly with 13C (U-13C palmitate) and 3-13C lactate or U-13C palmitate and 1-13C glucose or solely U-13C pyruvate and perfused for an additional 45 min. Hearts were freeze-clamped with Wollenberger tongs at the end of the experiment. Frozen hearts were powdered, extracted in 6% perchloric acid as previously described [23] and reconstituted in deuterated phosphate buffer (pH 6.8).
13C NMR spectroscopy
High-resolution 1H decoupled 13C NMR spectra of cardiac extracts were acquired with a 9.4T superconducting magnet at 100.5 MHz interfaced with a Jeol ECP400 NMR spectrometer. 24,000 scans were collected with a 60° pulse and acquisition delay of 1.3 s. Relative contributions of 13C labelled palmitate, lactate, glucose and pyruvate to the total acetyl-CoA pool entering the TCA cycle were determined from the isotopomer analysis of glutamate [24] using software provided by Dr. Mark Jeffrey (TCAcalcTM, University of Texas Southwestern Medical Centre). The contribution of unlabelled substrate to oxidative metabolism was calculated as described previously [21]. Subsequently, the absolute oxidation rates of each of the labelled substrates was determined using the relative contributions of the substrates and MVO2 as described previously [25, 26].
Tissue triglyceride measurements
In a separate series of experiments, ventricular tissue was rapidly removed, freeze-clamped and lipids extracted by a modified method of Bligh and Dyer [27]. Triglyceride content was assayed using a Sigma kit (Cat No: TR0100).
Tissue glycogen content
Myocardial glycogen content was determined in powdered ventricular tissue (of both perfused and unperfused hearts) as glucose equivalents after digestion with 30% KOH, ethanol precipitation and acid hydrolysis of glycogen as described previously [28].
Enzyme assays
Myocardial MCAD activity was measured in powdered ventricular tissue by following the decrease in ferricinium ion absorbance as described by Lehman et al. [29]. Pyruvate dehydrogenase (PDH) activity was determined in ventricular homogenates as described previously [30].
Serum analysis
Serum was separated from blood collected at the time of sacrifice by centrifugation (3,000 rpm for 10 min at 4°C). Concentrations of triglyceride and free fatty acids were measured using kits from Sigma (Cat No: TR0100) and Roche (Cat No: 1 383 175), respectively, whilst that of glucose was determined spectrophotometrically [31]. Haematocrit was measured using an ABL 77 series blood–gas analyser (Radiometer, Copenhagen).
RNA extraction
Total RNA was extracted from ventricular tissue using a modified version of Ullrich’s method [32] with guanidium isothiocyanate and separated via caesium chloride density gradient centrifugation. The extracted RNA was re-suspended in 30 μl of 1% DEPC-water, and its concentration measured spectrophotometrically at 260 nm.
Primers
Nucleotide sequences for cardiac specific genes encoding rat calsequestrin 2 (CSQ2), muscle carnitine palmitoyl transferase 1 (mCPT1), pyruvate dehydrogenase kinase (PDK) isoforms 2 and 4, peroxisome proliferator-activated receptor α (PPARα), fatty acid translocase (CD36), medium chain acyl-CoA dehydrogenase (MCAD), insulin-sensitive glucose transporter (GLUT 4) and mitochondrial uncoupling proteins (UCP) 2 and 3 were obtained from GenBank. Primers for these sequences (Table 1) were designed using Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). BLAST search for each of the primers confirmed homologous binding to the desired mRNA of rat cardiac muscle.
Real-time PCR
Two micro grams of total RNA was reverse-transcribed (in a 20 μl reaction volume) into first strand cDNA using Omniscript Reverse transcriptase kit (Qiagen, Cat No: 205111) according to manufacturer’s instructions. Quantification of the transcript expression was performed (in triplicate) by real-time PCR using the Bio-Rad iCycler® IQ detection system. The reaction mixture contained 1 μl of cDNA, 1.5 μl (75 nM final concentration) of each of the forward and reverse primers, 10 μl of 2× ABsolute SYBR green fluorescein master mix (ABgene, Cat No: AB-1219/A) containing Taq polymerase and 6 μl of RNAse free water. The correlation between the number of PCR cycles required for the fluorescent signal to reach the threshold (Ct) and the amount of standard was linear over a 5-log range of RNA for all assays. Ct values for all transcripts were normalised to that of housekeeping gene CSQ2 [33] and the difference in transcript expression between control and hypertrophied hearts calculated using modified ΔΔCt method [34].
Western blot analysis
Protein extracts from ventricular tissue were prepared as described previously [35]. Samples containing 50 μg protein were separated on 10 or 12% SDS PAGE gels, transferred to nitrocellulose membranes and probed with antibodies specific to Atrial natriuretic factor (ANF) (1:1000 dilution, Santa Cruz Biotechnology Inc), CD36 (1:6000 dilution, monoclonal antibody, Abcam, Cambridge, UK), GLUT4 (1:5000 dilution, Santa Cruz Biotechnology Inc) and PPARα (1:1000 dilution, Santa Cruz Biotechnology Inc) [35–38]. After conjugation with appropriate secondary antibodies (for ANF, goat anti-rabbit Horse radish peroxidase (HRP) 1:5000; for CD36, rabbit anti-mouse IgG HRP, 1:8000 dilution; for GLUT 4, donkey anti-goat IgG HRP 1:7000 and for PPARα, goat anti-rabbit HRP 1: 500), proteins were visualised using a ECL light detection kit (Amersham). Protein bands were quantified using commercially available software.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Single comparisons of means were performed by independent t-test using SPSS statistical software (v 11.5). A value of P < 0.05 was considered significant.
Results
Morphological characteristics of aortic constriction
Changes in wet heart weight, body weight and tibia length in animals undergoing aortic constriction are summarised in Table 2. Nine weeks post surgery, a marked increase in heart weight and heart weight-to-tibia length ratio—both indices of cardiac hypertrophy was observed in rats with aortic constriction (AC) compared to sham-operated controls (Con). However, body weights were comparable between the two groups. AC had no overt signs of heart failure, anaemia (haematocrit: 36.4 ± 1.3% AC vs. 35.4 ± 1.3% Con, N = 7) or fluid retention (%H2O) in the lungs (79.5 ± 0.3% AC vs. 79.2 ± 0.2% Con, N = 10). Indication of fluid/water retention in the lungs was determined by ascertaining the wet weight of the lungs and then, after drying at 40°C until constant weight, the dry wt. Serum glucose, triglycerides and free fatty acid levels were unchanged between the two groups (Table 3).
In vitro cardiac function
Cardiac function, MVO2 and efficiency, averaged over 45-min of perfusion with 13C enriched substrates are given in Table 4. Function was similar in both groups indicating that the hypertrophy was compensatory. AC consumed approximately 22% more oxygen than Con and hence showed a 19% decline in cardiac efficiency (P < 0.05).
Substrate utilisation
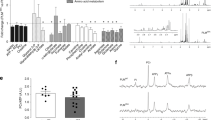
The absolute oxidation rates of 13C labelled palmitate, lactate, glucose and pyruvate are shown in Fig. 1. Palmitate oxidation was significantly reduced in AC relative to Con (20 ± 1.8 vs. 30 ± 2 nmoles/min/g; P < 0.05) whilst that of pyruvate was appreciably increased (57 ± 4 vs. 37 ± 3 nmoles/min/g; P < 0.01). In contrast, the oxidation rates of lactate (220 ± 10 AC vs. 202 ± 10 Con nmoles/min/g) and glucose (37 ± 2 AC vs. 38 ± 2 Con nmoles/min/g) were unaltered between the two groups. Thus, the shortfall in myocardial substrate provision in AC was met through an increased oxidation of pyruvate (54%).

Absolute oxidation rates of exogenous substrates. Oxidation rates of palmitate, lactate, glucose and pyruvate in Control (Con) and aortic constricted (AC) hearts. N = number of animals; *P < 0.05 AC versus Con. Open bars, Con; filled bars, AC
Metabolic gene expression
The mRNA transcript levels of the key metabolic enzymes are shown in Table 5. Nine weeks post surgery, the expression of PPARα and PPARα-regulated genes namely mCPT1, MCAD, UCP3 and PDK4 were significantly down-regulated in AC compared to Con. In addition, there was a marked reduction in PDK2 transcript levels. UCP2 expression was substantially higher in AC versus Con. However, no changes in the mRNA levels of CAT, CD36 and GLUT4 were observed.
Protein expression of ANF, CD36, GLUT4 and PPARα
Ventricular ANF, a phenotypic marker of hypertrophy was markedly elevated in AC 9 weeks post surgery (data not shown) and consistent with the morphological changes observed (Table 2). However, CD36 and GLUT4 levels were unaltered (Fig. 2) and consistent with the profile observed in transcript expression. Densitometric analysis revealed a significant decrease in PPARα protein levels in AC (Fig. 2, 0.088 ± 0.004 AC vs. 0.109 ± 0.0014, P < 0.0033, N = 4).

Protein expression of CD36, GLUT4 and PPARα. Western blots from representative gel of a Control (Con) and aortic constricted (AC) rat ventricular tissue run in duplicate. N = number of animals
Cardiac triglyceride concentrations
Myocardial triglyceride levels were not significantly different in AC compared to Con (1.83 ± 0.32 AC vs. 1.51 ± 0.20 Con, N = 3, μmoles/g wet heart wt).
Cardiac glycogen concentrations
Myocardial glycogen content was not significantly different in AC compared to Con in either perfused (21.3 ± 4.4 AC vs. 19.8 ± 4.6 Con, N = 5, μmoles/g wet heart wt) or unperfused hearts (12.1 ± 3.8 AC vs. 10.6 ± 3.9 Con, N = 2, μmoles/g wet heart wt).
Activity of key mitochondrial enzymes
MCAD activity, a marker of mitochondrial β-oxidative capacity was reduced by 19% (P < 0.05) in the hypertrophied myocardium compared to controls (5.2 ± 0.11 AC vs. 6.4 ± 0.32 Con, N = 4, μmoles/min/g wet heart wt). The proportion of active to total PDH (PDHa/PDHt) was unaltered in the hypertrophied hearts (0.27 ± 0.03 AC vs. 0.25 ± 0.01 Con, N = 3, μmoles/min/g wet heart wt).
Discussion
This study has demonstrated significant metabolic remodelling in the model of compensated cardiac hypertrophy employed here, highlighted by a marked decline in palmitate oxidation with a concomitant repression of PPARα and FAO genes and a substantial increase in pyruvate oxidation. The alteration in the profile of substrate oxidation is underpinned by molecular changes even during the compensatory phase of hypertrophy. To our knowledge, this is the first study to investigate the contribution of a physiological mixture of substrates to myocardial oxidative metabolism in compensated pressure-overload hypertrophy induced by aortic constriction.
Cardiac hypertrophy and function
Abdominal aortic constriction generated a moderate degree of cardiac hypertrophy, evident from the 22% increase in heart weight and heart weight-to-tibia length ratio (Table 2) with no evidence of heart failure. The degree of hypertrophy was comparable to previous observations using the same model [19, 30] and in models of supra-renal aortic constriction [39]. Re-expression of ANF in the ventricular tissue (a phenotypic marker of hypertrophy) further supported the morphological findings. Hypertrophy was compensatory in nature as reflected by the preserved cardiac function determined by rate pressure product. Nevertheless, cardiac efficiency was somewhat compromised as a result of enhanced myocardial oxygen consumption suggesting that the hypertrophied hearts can extract more oxygen despite a lower flow rate (Table 4). Impaired contractile performance, increased energy utilisation for excitation–contraction coupling, elevated basal metabolic rate or changes in substrate supply can all result in cardiac inefficiency [40, 41]. Given that both substrate supply and contractile performance were maintained here and basal metabolic rate was unlikely to be affected (as hearts were perfused at a constant flow rate), one possibility for this decline in cardiac efficiency could be the high energy cost of excitation–contraction coupling resulting from abnormal Ca2+ homeostasis in the hypertrophied myocardium [42]. In contrast, earlier studies have shown that MVO2 was either unchanged [43] or reduced [44] in the hypertrophied myocardium of spontaneously hypertensive rats (SHR) and could be attributed to the disparity in the experimental models (genetic model of hypertension vs. one induced by aortic constriction).
Profile of energy provision
Nine weeks post aortic constriction, a significant reduction in palmitate oxidation (33%) was observed in hypertrophied hearts compared to controls (Fig. 1). This finding is consistent with previous studies showing a reduction in long-chain FAO in rat hearts subjected either to a more severe pressure [10, 11] or volume [12] overload. In the present study, the decline in FAO may be attributed to altered substrate uptake, changes in metabolic gene expression or potentially myocardial carnitine depletion. Data here show that CD36 expression is unchanged in the hypertrophied hearts (Fig. 2) implying that alterations in FA uptake are unlikely. Indirect support for this comes from the myocardial triglyceride content which is also unaltered in the hypertrophied myocardium. Consequently, down-regulation of PPARα and FAO genes (CPT1 and MCAD) is more likely to be responsible for this decline in palmitate oxidation in AC. This view is strengthened by the observed reduction in PPARα protein expression (Fig. 2) and MCAD activity in the hypertrophied myocardium. Further, left ventricular hypertrophy has been associated with a decrease in myocardial carnitine (an essential cofactor that transports long-chain FA across the mitochondrial membrane) levels [10–12]. Therefore, low carnitine levels may contribute to the reduced palmitate oxidation observed here.
The decline in palmitate oxidation was associated with a concomitant increase in pyruvate oxidation in AC. Enhanced mitochondrial pyruvate oxidation has been shown to augment contractile function in isolated hearts, primarily by increasing sarcoplasmic reticular Ca2+ turnover [15]. Consequently, the increased reliance of the hypertrophied myocardium on pyruvate may account for the preserved contractile function observed here. Elevated pyruvate use could be attributed to the fact that in the present study, a more physiological mixture of substrates was provided to the hearts including both lactate and pyruvate as alternative carbohydrate sources, whereas previous studies used solely glucose (11 mM) and palmitate (0.4 or 1.2 mM) as substrates [11, 12]. In contrast, pyruvate oxidation was unaltered in the hypertrophied myocardium of SHRs in studies by Des Rosiers and colleagues perfused in the presence of a physiological mixture of substrates [43, 44]. Differences in the model [i.e. genetic hypertension (SHR) vs. surgical induction of hypertension (aortic constriction)] and heart perfusion system (working vs. isovolumic) may account for the observed disparity in pyruvate oxidation.
In the present study, myocardial efficiency declined despite a favourable shift in substrate preference from FA towards pyruvate (an oxygen efficient fuel). This could be attributed to oxygen wasting in the hypertrophied myocardium via FA-induced uncoupling of oxidative phosphorylation [45] and/or oxygen dependent cycling of FA into triglycerides and vice versa [22], a consequence of elevated myocardial FA levels due to reduced FAO with preserved FA uptake.
In contrast to the findings here, earlier studies have demonstrated increased glucose utilisation in the hypertrophied myocardium to compensate for the decrease in FAO [10–12]. However, as mentioned earlier, these studies employed either solely glucose (11 mM) and palmitate (0.4 or 1.2 mM) [11, 12] or glucose (11 mM), palmitate (0.4 mM) and lactate (0.5 mM) [10] as substrates, thereby excluding pyruvate. In addition, perfusing the hearts in the isovolumic mode as opposed to the working mode may account for this discrepancy in the profile of substrate oxidation. Preference was given to a non-circulating isovolumic mode here in order to avoid ambiguity arising from the re-entry of 13C label into the system and avert functional deterioration from toxic metabolites [46].
Further, the present model of cardiac hypertrophy differs from previous models with respect to age of animals at the time of experiment, strain of animals used and severity of hypertrophy induced which may also explain the differences in the profile of substrate use. Allard and colleagues performed aortic constriction on 3-week old (juvenile) Wistar–Kyoto [10] and Sprague–Dawley [39] rats whereas adult Sprague–Dawley rats (7–8 week) were used in this study. Christe and Rodgers investigated substrate oxidation in SHRs [11]. Of interest in the present study was the finding that the oxidation rate of lactate was substantial and similar in both control and hypertrophied hearts, highlighting again the appreciable contribution that lactate makes to cardiac energy production [21, 47].
Previous studies have shown significant glycogen turnover and contribution to energy production in both control and hypertrophied hearts [39, 48]. However, in the present study, there was no alteration in glycogen content in the perfused myocardium (in either group) implying that the contribution of glycogen to oxidative metabolism was unchanged in the face of hypertrophy.
Metabolic gene expression
In parallel with the changes in myocardial substrate oxidation, real-time PCR analysis revealed a marked reduction in the transcript expression of PPARα and PPARα-regulated genes (mCPT1, MCAD, UCP3 and PDK4) in pressure-overloaded hearts. This pattern is consistent with the down-regulation of the adult phenotype observed in cardiac hypertrophy [16, 18, 49]. However, the model of hypertrophy and the stage of elucidation of gene expression were different in these studies. In contrast, expression of PPARα, mCPT1, UCP3 and PDK4 was unaltered in the compensated hypertrophied heart in a recent study [50]. Nevertheless, these authors performed supra-renal aortic constriction in Wistar rats (as opposed to inter-renal aortic constriction in Sprague–Dawley rats in the present study) which may explain the dissimilarities in the profile of gene expression.
The importance of PPARα in the control of cardiac FA metabolism has been demonstrated in a wide variety of studies: from the use of PPARα null mice [where the expression of myocardial genes encoding FA transport and oxidation enzymes is diminished] [51] to reduced transcript expression and DNA binding capacity of PPARα in parallel with decreased expression of FAO genes [16] and down-regulation of the PPARα/PGC1 complex [52] in pathologic hypertrophy. In the light of these studies, down-regulation of PPARα transcript (Table 5) and protein expression (Fig. 2) may be responsible for the decline in the expression of FAO genes observed here. Young and colleagues have shown that reactivation of PPARα in the hypertrophied hearts can result in severe contractile dysfunction [53]. Repression of PPARα can therefore be considered an adaptive response in compensated hypertrophy. Down-regulation of mCPT1 may reflect an overall reduction in carnitine-dependent FA transport and in consequence, diminished FAO in cardiac hypertrophy [12].
Despite the reduction in PDK4 expression at the RNA level, no change in the proportion of PDH in the active form was observed. Given that regulation of PDH activity involves a number of different mechanisms in addition to phosphorylation/dephosphorylation, it is possible that changes in effector ratios such as mitochondrial NADH/NAD+ or acetylCoA/CoA might modulate activity at this compensated phase of hypertrophy. An alternative explanation for the discrepancy between PDK4 transcript expression and the proportion of PDH in the active form may result from the timing of tissue harvesting. Expression of PDK4 shows a diurnal oscillation at the mRNA level [54] and it is possible that the time of sacrifice did not correspond to the peak for mRNA levels.
The role of UCPs in the heart is still unclear. In adipose tissue and skeletal muscle, UCPs are known to dissipate the proton electrochemical gradient formed during mitochondrial respiration and generate heat instead of ATP [55]. It has been proposed that UCP3 may regulate cardiac FA utilisation and ATP synthesis [56] and that its expression declines when the hypertrophied heart attempts to maintain function in response to altered workload [49]. Repression of UCP3 with a concomitant decline in FAO observed here is consistent with this view. In addition, down-regulation of UCP3 in parallel with PPARα supports that this transcription factor is regulated by PPARα [49, 55]. However, the up-regulation of UCP2 observed here may contribute to uncoupling of mitochondrial oxidative phosphorylation and thus account for the higher oxygen consumption and lower cardiac efficiency. Pressure-overload hypertrophy has been associated with repression of both UCP 2 and 3 mRNA levels in a previous study [49]. Differences in the model of hypertrophy, extent of hypertrophy and the stage of investigation could account for these discrepancies.
The expression profile of CD36—a key transporter of LCFA across the cardiac plasma membrane—is largely unknown in compensated hypertrophied hearts. Findings here indicate that mRNA and protein levels of CD36 are unaltered in the hypertrophied ventricles (Table 5, Fig. 2) and thus the decline in palmitate oxidation is unlikely to occur as a result of restricted FA uptake. This is in agreement with a previous study where CD36 transcript expression was unchanged in the hypertrophied ventricles following myocardial infarction [57].
Previous studies have shown that GLUT4 mRNA content is reduced in the failing rat hearts [18, 58] but unchanged in hypertrophied ventricles [18] suggesting that insulin-dependent glucose uptake is unaffected in the early phase of cardiac hypertrophy but insulin resistance is a later event in the progression to heart failure [58]. In the present study, unchanged GLUT4 mRNA and protein levels in AC (Table 5, Fig. 2) would support this view.
In conclusion, we have shown that expression of PPARα and PPARα-regulated genes are coordinately down-regulated in the early stage of cardiac hypertrophy with a concomitant decline in palmitate oxidation—highlighting the down-regulation of the adult phenotype. This is paralleled by an increased reliance on pyruvate and maintenance of cardiac function. Whether the decline in FAO and enhanced pyruvate use in the hypertrophied heart become maladaptive and contribute to the development of heart failure warrants further investigation.
Abbreviations
- ATP:
-
Adenosine triphosphate
- MCAD:
-
Medium-chain acyl-CoA dehydrogenase
- NMR:
-
Nuclear magnetic resonance
- PCR:
-
Polymerase chain reaction
- PDH:
-
Pyruvate dehydrogenase
- PPARα:
-
Peroxisome proliferator-activated receptor-α
- PGC1:
-
Peroxisome-proliferator-activated receptor-γ co-activator 1
References
Seymour AM (2003) Mitochondrial function—a limiting factor in heart failure? In: Dhalla NS, Rupp H (eds) Pathophysiology of cardiovascular disease, 1st edn. Kluwer academic publishers, Boston, pp 23–33
Frohlich ED, Apstein C, Chobanian AV, Devereux RB, Dustan HP, Dzau V, Fauad-Tarazi F, Horan MJ, Marcus M, Massie B (1992) The heart in hypertension. N Engl J Med 327:998–1008
Lehman JJ, Kelly DP (2002) Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail Rev 7:175–185
Hawkins NM, Wang D, McMurray JJ, Pfeffer MA, Swedberg K, Granger CB, Yusuf S, Pocock SJ, Ostergren J, Michelson EL, Dunn FG (2007) Prevalence and prognostic implications of electrocardiographic left ventricular hypertrophy in heart failure: evidence from the CHARM programme. Heart 93:59–64
Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr, Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J, Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-Smoller S, Wilson M, Wolf P (2006) Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 113:e85–e151
van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ (2004) Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res 61:218–226
Gerdes AM (2002) Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail 8:S264–S268
Burgess ML, Buggy J, Price RL, Abel FL, Terracio L, Samarel AM, Borg TK (1996) Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. Am J Physiol 270:H151–H159
Bers DM (2006) Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21:380–387
Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD (1994) Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 267:H742–H750
Christe ME, Rodgers RL (1994) Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 26:1371–1375
el Alaoui-Talibi Z, Guendouz A, Moravec M, Moravec J (1997) Control of oxidative metabolism in volume-overloaded rat hearts: effect of propionyl-L-carnitine. Am J Physiol 272:H1615–H1624
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Lloyd S, Brocks C, Chatham JC (2003) Differential modulation of glucose, lactate, and pyruvate oxidation by insulin and dichloroacetate in the rat heart. Am J Physiol Heart Circ Physiol 285:H163–H172
Mallet RT, Sun J (1999) Mitochondrial metabolism of pyruvate is required for its enhancement of cardiac function and energetics. Cardiovasc Res 42:149–161
Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP (1996) Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94:2837–2842
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104:2923–2931
Rosenblatt-Velin N, Montessuit C, Papageorgiou I, Terrand J, Lerch R (2001) Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc Res 52:407–416
Boateng S, Seymour AM, Dunn M, Yacoub M, Boheler K (1997) Inhibition of endogenous cardiac phosphatase activity and measurement of sarcoplasmic reticulum calcium uptake: a possible role of phospholamban phosphorylation in the hypertrophied myocardium. Biochem Biophys Res Commun 239:701–705
Yin FC, Spurgeon HA, Rakusan K, Weisfeldt ML, Lakatta EG (1982) Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am J Physiol 243:H941–H947
Sample J, Cleland JG, Seymour AM (2006) Metabolic remodeling in the aging heart. J Mol Cell Cardiol 40:56–63
How OJ, Aasum E, Kunnathu S, Severson DL, Myhre ES, Larsen TS (2005) Influence of substrate supply on cardiac efficiency, as measured by pressure-volume analysis in ex vivo mouse hearts. Am J Physiol Heart Circ Physiol 288:H2979–H2985
Seymour AM, Eldar H, Radda GK (1990) Hyperthyroidism results in increased glycolytic capacity in the rat heart. A 31P-NMR study. Biochim Biophys Acta 1055:107–116
Malloy CR, Sherry AD, Jeffrey FM (1990) Analysis of tricarboxylic acid cycle of the heart using 13C isotope isomers. Am J Physiol 259:H987–H995
Lloyd SG, Wang P, Zeng H, Chatham JC (2004) Impact of low-flow ischemia on substrate oxidation and glycolysis in the isolated perfused rat heart. Am J Physiol Heart Circ Physiol 287:H351–H362
Malloy CR, Jones JG, Jeffrey FM, Jessen ME, Sherry AD (1996) Contribution of various substrates to total citric acid cycle flux and anaplerosis as determined by 13C isotopomer analysis and O2 consumption in the heart. Magma 4:35–46
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Richardson S (2002) Studies of ischaemia and reperfusion in cardiac hypertrophy, PhD thesis. University of Hull, Hull, p 253
Lehman TC, Hale DE, Bhala A, Thorpe C (1990) An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal Biochem 186:280–284
Seymour AM, Chatham JC (1997) The effects of hypertrophy and diabetes on cardiac pyruvate dehydrogenase activity. J Mol Cell Cardiol 29:2771–2778
Trinder P (1969) Determination of blood glucose using an oxidase-peroxidase system with a non-carcinogenic chromogen. J Clin Pathol 22:158–161
Sambrook J, Russel DW (2001) Extraction, purification and analysis of mRNA from eukaryotic cells. 7.3–7.25
Boheler KR, Volkova M, Morrell C, Garg R, Zhu Y, Margulies K, Seymour AM Lakatta EG (2003) Sex- and age-dependent human transcriptome variability: implications for chronic heart failure. Proc Natl Acad Sci U S A 100:2754–2759
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Boateng SY, Seymour AM, Bhutta NS, Dunn MJ, Yacoub MH, Boheler KR (1998) Sub-antihypertensive doses of ramipril normalize sarcoplasmic reticulum calcium ATPase expression and function following cardiac hypertrophy in rats. J Mol Cell Cardiol 30:2683–2694
Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN (2000) Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem 275:14501–14508
Santalucia T, Camps M, Castello A, Munoz P, Nuel A, Testar X, Palacin M, Zorzano A (1992) Developmental regulation of GLUT-1 (erythroid/Hep G2) and GLUT-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 130:837–846
Morgan EE, Chandler MP, Young ME, McElfresh TA, Kung TA, Rennison JH, Tserng KY, Hoit BD, Stanley WC (2006) Dissociation between gene and protein expression of metabolic enzymes in a rodent model of heart failure. Eur J Heart Fail 8:687–693
Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD (1997) Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation 96:676–682
Mjos OD (1971) Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs. J Clin Invest 50:1386–1389
Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, Gerstenblith G (1991) Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Physiol 261:H741–H750
Houser SR, Piacentino V, Weisser J (2000) Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 32:1595–1607
Vincent G, Khairallah M, Bouchard B, Des Rosiers C (2003) Metabolic phenotyping of the diseased rat heart using 13C-substrates and ex vivo perfusion in the working mode. Mol Cell Biochem 242:89–99
Labarthe F, Khairallah M, Bouchard B, Stanley WC, Des Rosiers C (2005) Fatty acid oxidation and its impact on response of spontaneously hypertensive rat hearts to an adrenergic stress: benefits of a medium-chain fatty acid. Am J Physiol Heart Circ Physiol 288:H1425–H1436
Borst P, Loos JA, Christ EJ, Slater EC (1962) Uncoupling activity of long-chain fatty acids. Biochim Biophys Acta 62:509–518
Garlick PB, Mashiter GD, Di Marzo V, Tippins JR, Morris HR, Maisey MN (1989) The synthesis, release and action of leukotrienes in the isolated, unstimulated, buffer-perfused rat heart. J Mol Cell Cardiol 21:1101–1110
Drake AJ, Haines JR, Noble MI (1980) Preferential uptake of lactate by the normal myocardium in dogs. Cardiovasc Res 14:65–72
Goodwin GW, Arteaga JR, Taegtmeyer H (1995) Glycogen turnover in the isolated working rat heart. J Biol Chem 270:9234–9240
Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H (2001) Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. Faseb J 15:833–845
Degens H, de Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ, van der Vusse GJ, van Bilsen M (2006) Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol 101:17–26
Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, Kelly DP (1998) A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor alpha-deficient mice. J Clin Invest 102:1083–1091
Sack MN, Disch DL, Rockman HA, Kelly DP (1997) A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci U S A 94:6438–6443
Young ME, Laws FA, Goodwin GW, Taegtmeyer H (2001) Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem 276:44390–44395
Young ME (2006) The circadian clock within the heart: potential influence on myocardial gene expression, metabolism and function. Am J Physiol Heart Circ Physiol 290:H1–H16
Boss O, Hagen T, Lowell BB (2000) Uncoupling proteins 2 and 3: potential regulators of mitochondrial energy metabolism. Diabetes 49:143–156
Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K (2001) Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am J Physiol Heart Circ Physiol 280:H977–H983
Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP (2005) Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. Febs J 272:2684–2695
Paternostro G, Clarke K, Heath J, Seymour AM, Radda GK (1995) Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc Res 30:205–211
Acknowledgements
Ashwin Akki was a recipient of the Graduate Teaching Assistantship from the University of Hull. We thank Mrs. Jenny Foster and Mrs. Kath Bulmer for excellent technical support, Mr. Denys Bilko for technical advice with Real-time PCR and Mr. K.Y. Lee for help with Western blotting. Thanks also go to Dr. Jessica Sample for constructive discussions and critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akki, A., Smith, K. & Seymour, AM.L. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem 311, 215–224 (2008). https://doi.org/10.1007/s11010-008-9711-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9711-y