
Abstract
Chronic cardiac ischemia/hypoxia induces coronary collateral formation and cardiomyocyte proliferation. Hypoxia can induce cellular adaptive responses, such as synthesis of VEGF for angiogenesis and IGF-2 for proliferation. Both reduce apoptotic effects to minimize injury or damage. To investigate the mechanism of neoangiogenesis and proliferation of fetal heart under umbilical cord compression situation, we used H9c2 cardiomyoblast cell culture, and in vivo embryonic hearts as our study models. Results showed hypoxia induced not only the increase of IGF-2 and VEGF expression but also the activation of their upstream regulatory genes, HIF-1α and Shh. The relationship between HIF-1α and Shh was further studied by using cyclopamine and 2-ME2, inhibitor of Shh and HIF-1α signaling, respectively, in the cardiomyoblast cell culture under hypoxia. We found that the two inhibitors not only blocked their own signal pathway, but also inhibited each other. The observations revealed when fetal heart under hypoxia that HIF-1α and Shh pathways maybe involve in cell proliferation and neoangiogenesis to minimize injury or damage, whereas the complex cross-talk between the two pathways remains unknown.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic injury to the myocardium due to the spectrum of coronary occlusion events known as acute coronary syndromes is a large proportion of all causes of death in western society [1]. Also a pregnancy mother exposed to both tobacco and alcohol shows ischemia injury to infant. For example, maternal nicotine injection during late pregnancy in sheep increased fetal blood pressure and umbilical vascular resistance and decreased the fetal heart rate and umbilical blood flow, even consequently vascular disruption seen in this infant [2, 3]. A greater understanding of the cellular and molecular mechanisms involved in ischemic injury may foster additional improvements in clinical care.
Ischemic injury results in deprivation of oxygen and nutrients; the growth and viability of cells is reduced. Hypoxia-inducible factor (HIF)-1α helps to restore oxygen homeostasis by inducing glycolysis, erythropoiesis, and angiogenesis [4]. Complex cellular and systemic regulatory mechanisms control O2 homeostasis. The key regulator of O2 homeostasis is HIF-1α, a basic-helix-loop-helix-PAS transcription factor [1]. HIF-1α activates transcription of genes whose protein products increase O2 availability or promote metabolic adaptation to hypoxia including erythropoietin and VEGF, which control erythropoiesis and angiogenesis, respectively [5]. HIF-1α also plays an important role in cellular proliferation. Several previous studies support that a strong correlation between HIF-1α overexpression and cellular proliferation [6, 7]. In normoxia, HIF-1α also can be induced via activated PI3K by some growth factors and cytokines, including insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF), interleukin-1 (IL-1) [8–10]. IGF-1 or IGF-2 can induce HIF-1α protein expression, and interestingly HIF-1α was required for expression of genes encoding IGF-2, IGFBP-2, and IGFBP-3; these establish a positive reciprocal relationship between the HIF-1 and IGF systems that has important implications for the understanding of cellular energy metabolism, growth control, and tumor progression [5].
Insulin-like growth factors (IGFs) are small peptides, which are closely related to proinsulin. IGF-1 and IGF-2 modulate cell proliferation and differentiation [11] during embryogenesis through paracrine [12] or autocrine [13] pathways. In vitro studies, using 3T3 cells, have demonstrated that IGF-1R is responsible for the determination of whether cells progress along a mitogenic pathway or undergo apoptosis [14], and both IGF-1 and IGF-2 act via IGF-1R for mitogenic signaling in murine embryonic development [15]. IGF-1 and IGF-2 are known potent angiogenic factors in a murine retinal model [16].
Hedgehog (Hh) proteins are morphogens regulating epithelial–mesenchymal signaling during several crucial processes of embryonic development, including muscle patterning. Sonic (Shh), Indian (Ihh), and Desert (Dhh) hedgehog constitute the repertoire of Hh genes in mammalian. The activities of all three are transduced via the Patched (Ptc1) receptor [17]. In addition, Shh stimulates fibroblasts in vitro to produce a combination of potent angiogenic factors, including the three major isoforms of VEGF, Ang-1, and Ang-2. Shh seems to act as an indirect angiogenic agent and may trigger neovascularization through Shh/Ptc1 signaling specifically in mesenchymal cells [18]. Recently observations indicate that exogenous administration of Shh induces angiogenesis and plays a physiological role in muscle regeneration after ischemia in adults [17].
Taken together, cardioprotective effects were induced when ischemia occurred; some protective proteins such as VEGF can induce anti-apoptotic protein and improve endothelial cell survival and angiogenesis; HIF-1α regulates some proteins adapted to hypoxia, including IGF-2 and VEGF; IGF-2 improves growth and proliferation; Shh improves myogenesis and synergistic with IGF. We suppose that in embryo cardiac ischemia, HIF-1α and Shh could be induced and signaling to the downstream target genes VEGF and IGF-2 to exert cardiac protective function.
Materials and methods
Cell culture
The rat cardiomyoblast cell line H9c2 was maintained in DMEM media (Sigma, St. Louis, USA) supplemented with 10% fetal bovine serum (Hyclone, Utah, USA), 1.5 g/l sodium bicarbonate, and 1% antibiotic–antimycotic (Invitrogen, California, USA) in an atmosphere of 95% O2–5% CO2 at 37°C. For experiments, all hypoxia treatments consisted of about 5% O2 in serum-starvation (1% FBS) or normal condition DMEM in an air-tight humidified chamber at 37°C as described previously.
Western blotting
During cell harvesting, proteins were extracted using lysis buffer (50 mM Tris–base, 0.5 M NaCl, 1.0 mM EDTA, 1% NP40, 1% Glycerol, 1 mM β-Mercaptoethanol, Proteinase k inhibitor) and centrifuged to discard the pellet. The protein concentrations were determined by the Lorry assay. The proteins of cell lysates were analyzed by SDS-PAGE, and proteins were transferred to nitrocellulose (or PVDF). Residual protein sites on nitrocellulose papers were incubated at room temperature for 2 h and then immersed in blocking buffer containing 5% skim milk in Tween/Tris–buffer saline (TBS). After washing with buffer, the nitrocellulose papers were immersed in monoclonal antibodies of HIF-1α, Shh, IGF-1(R)/2(R), VEGF, α-tubulin (Santa Cruz, California, USA) diluted with TBS containing 5% skim milk and incubated at room temperature for 4 h. The immunoblots were then washed in triplicate with TBS for 10 min before immersing in the second antibody solution for 1 h and diluted 1000-folds in TBS. The filters were then washed in triplicate with TBS for 10 min. Antibody reaction was visualized with chemiluminescent reagent for Western blotting.
RT-PCR and PCR
The RNA was extracted from the H9c2 cells with hypoxia treatment. The cDNA of the cells was amplified by PCR with Shh primers: forward primer (5′-GGCAG ATATG AAGGG AAGAT-3′) and reverse primer (5′-ACTGC TCGAC CCTCA TAGTG-3′); IGF-1 primers: forward primer (5′-TGGAT GCTCT TCAGT TCGTG-3′) and reverse primer (5′-CAACA CTCAT CCACA ATGCC-3′); IGF-1R primers: forward primer (5′-CCTAG ACAAC CAGAA CTTGC-3′) and reverse primer (5′-GTCTA CATCC ACCAT GTTCC-3′); IGF-2 primers: forward primer (5′-CTTGT TGACA CGCTT CAGT-3′) and reverse primer (5′-GTTCA CTGAT GGTTG CTGGA-3′); IGF-2R primers: forward primer (5′-GCAAG GGCAT AAGGT GAA-3′) and reverse primer (5′-TGTAA GTCAC CCTGT GCAA-3′); VEGF primers: forward primer (5′-CTTCC TACAG CACAG CAGAT GTGAA-3′) and reverse primer (5′-TGGTG ACATG GTTAA TCGGT CTTTC-3′). Aliquots of the PCR products were electrophoresed on 1.5% (w/v) agarose gels stained with Gelstar and photographed.
Immunohistochemistry
Fix embryo with 4% paraformaldehyde in PBS (pH 7.4). Dehydrate and embed specimen in wax, and section specimen to a thickness of 5–7 μm using a microtome. Coat slides with 3-triethoxysilylpropylamine (TESPA; Sigma, USA) to increase adhesion of the sections. Collect sections on a glass slide by placing a drop of water on the slide and positioning sections on this drop, and place the slides on slides warmer (∼47°C) until moisture is no longer visible. Allow to air-dry for 1–2 days before staining. Dewax sections by immersing slides in holder in staining jars containing the following series: xylene, xylene, 100% ethanol, 100% ethanol, 90% ethanol, 70% ethanol, 50% ethanol, 30% ethanol, TM (10 mM Tris–HCl, 100 mM MgCl2), and ddH2O, each immersing for 3–5 min. Immerse slides in a holder container with citric acid buffer (0.01 M citric acid, 0.2% NP-40, pH 6.0) and boil for three to five times. Immerse the slides in blocking buffer (10 mM Tris–HCl, 100 mM MgCl2, 0.1% Tween 20, 1% BSA, 5% FBS), and incubate at room temperature for 1 h. Wipe the back and edges of the slide with 3MM filter paper, and cover the sections with diluted primary antibody and incubate in the humidity box at room temperature overnight. Wash the slides twice in 500 ml of TBST (10 mM Tris–HCl, 150 mM NaCl, 0.1% Tween 20) in a staining jar at room temperature for 30 min each time. Wipe the back and edges of the slide with 3MM filter paper, add secondary antibody, and incubate in the humidity box at room temperature for 1 h. Wash the slides immersed in TBST twice for 10 min. Immerse slides in the staining jar with alkaline phosphatase enzyme substrate buffer, containing NBT/BCIP (Roche, USA) and NTMT buffer (100 mM NaCl, 100 mM Tris–HCl, 5 mM MgCl2) for color development. Stop enzyme reaction by immersing slide in stop buffer, and mount the slides in mounting solution.
Surgical procedure for maternal–fetal blood flow obstruction
The transient ischemic model developed for fetal rats by Magal et al. [19] was adapted to the pregnant mouse. On the 12.5 days of gestation (E12.5) of ICR mice, pregnant mice were anesthetized by intramuscular injection of pentobarbital. A low abdominal midline incision was made and the uterine horns were exposed. Two delicate artery clamps were used to occlude the uterine vessels near the lower and upper ends of the right ipsilateral horn. After 7 min [20], clamps were removed and the exposed horns were returned back to the peritoneal cavity for 6 h of reperfusion. The animals were sacrificed and hearts were immediately excised for RT-PCR or immunohistochemistry. The other uterine was not subjected to blood vessels occlusion served as control.
Results
Ischemia induced HIF-1α, VEGF, IGF-1/1R, IGF-2/2R, Shh gene expression in embryonic heart
The mRNA levels of HIF-1α, VEGF, IGF-1/1R, IGF-2/2R, and Shh all increased significantly in ischemia treatment of embryonic heart (Fig. 1). VEGF and IGF-2, those downstream genes of HIF-1α, increased simultaneously. Also, IGF-1/1R as survival signal increased after ischemia/reperfusion. Shh, an indirect inducer of VEGF and IGF-2, also increased significantly.

mRNA expression in mouse embryonic heart ischemia. The uterine vessels were occluded leading to ischemia of mouse embryo (E12.5) for 7 min, then released for 6 h of reperfusion. One of two uteri was not occluded served as control. Embryonic hearts were drawn out and analyzed by RT-PCR. The mRNA levels of HIF-1α, Shh, VEGF, IGF-2/2R, and IGF-1/1R were significantly higher after ischemia treatment. C (control); I (ischemia); HIF-1α (hypoxia-inducible factor-1α); Shh (sonic hedgehog); VEGF (vascular endothelial growth factor). The histogram was showed below
Ischemia induced HIF-1α, VEGF, IGF-1/1R, IGF-2/2R, Shh protein expression in embryonic heart
Through transient occlusion of uterine vessels leading to ischemia in embryonic hearts, in IHC data, we demonstrated HIF-1α, VEGF, IGF-1/1R, IGF-2/2R, and Shh protein levels were higher indeed, especially in atrium, ventricle wall, and even in vessel walls (Fig. 2). Shh protein expressed in left ventricle and aorta after ischemia/reperfusion treatment (Fig. 2b, d, arrow head). HIF-1α and VEGF expressed in left ventricle and pulmonary vessels (Fig. 2f, h, j, l, arrow head). IGF-2/2R and IGF-1/1R expressed obviously in heart area, especially in left atrium and left ventricle (Fig. 2n, p, r, t, v). All these genes not only increased simultaneously, but also are expressed colocally. We speculate these proteins exert functions to improve angiogenesis or proliferation in embryonic heart when ischemia occurred in vivo and have somewhat a relationship between each other.

Pattern of protein expression in mouse embryonic heart ischemia. The uterine vessels were occluded leading to ischemia of mouse embryo (E12.5) for 7 min, then released for 6 h of reperfusion. One of two uteri was not occluded served as control. Embryonic hearts were drawn out for tissue section and analyzed by immunohistochemistry. Shh (b, d), HIF-1α (f, h), VEGF (j, l), IGF-2 (n, p), IGF-2R (r), IGF-1 (t), and IGF-1R (v) protein levels were higher at heart atrium, ventricle, and artery wall after ischemia. All sections were transverse. Larger visions of the first row were showed in the second row of the panel. PA (pulmonary artery); LV (left ventricle); LA (left atrium); A (aorta)
Hypoxia, serum starvation, and ischemia-induced mRNA and protein expression in cardiomyoblast (H9C2)
To confirm our observation in vivo, we used cardiomyoblast cell line (H9C2) treating with hypoxia (<5% O2), serum starvation (1% FBS), and hypoxia plus serum starvation which mimic ischemia. The data show mRNA and protein levels of HIF-1α were significantly increased in hypoxia condition and become higher with increase in time (Fig. 3a, b). The mRNA level of Shh showed increase with time extension in all three conditions; however in the protein level, change of precursor form was similar as mRNA change; the active form of Shh was poor production as a secreted protein (Fig. 3a, b). VEGF also present a more sensitive response to hypoxia than the other two conditions (Fig. 3a). IGF-2 was more influenced in serum starvation and ischemia conditions similar to Shh-expressed pattern (Fig. 3b). These results suggest that ischemia-induced protein expression maybe leading to angiogenesis or proliferation could be reproved in cardiomyoblast performance a new observation for clinical application.

Hypoxia, serum starvation, and ischemia-induced mRNA and protein expression in cardiomyoblast (H9C2). Cells were treated with serum starvation (1% FBS), hypoxia (<5% O2), or ischemia, and cultured for 6, 12, or 18 h, then analyzed by RT-PCR (a) or Western blot (b). HIF-1α mRNA and protein levels were significantly induced in hypoxia and ischemia conditions, and VEGF, the downstream gene of HIF-1α, also showed higher mRNA expression simultaneously (a, b). Protein and mRNA levels of Shh were significantly higher in serum starvation, hypoxia, and ischemia conditions, and the downstream gene, IGF-2, also showed higher protein level (a, b). C (control); S (serum starvation); H (hypoxia); HS (hypoxia plus serum starvation as ischemia). The histogram was showed below
HIF-1α and Shh signaling pathways might have complex cross-talk in hypoxia condition
Cardiomyoblast (H9C2) cells were treated with cyclopamine, a steroidal alkaloid that blocks the intracellular Shh signaling [21], and 2-ME2 that blocks HIF-1α signaling in hypoxia condition for 3 or 6 h. The results show that the mRNA expression of IGF-2, Ptch-1, and HIF-1α was reduced significantly only by 5 μM cyclopamine, but insignificantly affected by 1 or 10 μM (Fig. 4a). When blocking HIF-1α signaling, leading to HIF-1α protein degradation and the downstream gene, VEGF, also decreased protein production and simultaneously decreased Shh protein production (Fig. 4b). These results indicate they might influence the signal system between each other that improves cardiomyoblast proliferation and angiogenesis leading to cardiac protection.

HIF-1α and Shh signaling pathways might have complex cross-talk in hypoxia condition. Cardiomyoblast cell line (H9c2) were treated with cyclopamine (Shh signal inhibitor) or 2-ME2 (2-Methoxyestradiol; HIF-1α signal inhibitor) and incubated in hypoxia (<5% O2) condition for 3 or 6 h, analyzed by RT-PCR (a), or Western blot (b). Blocking Shh signaling pathway resulted in decreased mRNA levels of Ptch-1, IGF-2, and influence of HIF-1α mRNA expression (a). Blocking HIF-1α signaling pathway resulted in decreased protein levels of VEGF and Shh (b). The histogram was showed below
Discussion
Ischemia occurred in embryo heart conditions such as uterine hypertonicity, abruption placentae, and umbilical cord compression may cause fetal hypoxia which, if sustained, can cause fetal death [20]. Ischemia heart diseases also cause heart failure in adult. In past few years, studies of the cellular and molecular mechanism for protecting heart function after ischemia and reperfusion injury had progressed rapidly. We try to figure out the molecular mechanism and the signaling pathway induced by ischemia in fetal rat heart for clinical application or gene therapy. The conclusions were supported by studies demonstrating that transient ischemia in embryo induced expression of HIF-1α, Shh, IGF-2, and VEGF genes in heart. Additionally in vitro study, exposure of cultured cardiomyoblast cells to hypoxia and serum starvation also induced expression of HIF-1α, Shh, IGF-2, and VEGF genes, which suggests that these proteins improve the cardiac protective effect especially in cardiomyoblast. In this article, we report that exposure of cells to HIF-1α and Shh inhibitors, respectively, resulted in the inhibition of each other. These results indicate there might have been complex cross-talk between the HIF-1α and Shh systems that has important implications for the understanding of proliferation and angiogenesis after ischemia.
In our research, serum starvation and hypoxia regulate different pathways in cardiomyoblast; there was no HIF-1α expression in serum starvation condition, and induced Shh and IGF-2 instead obviously. Only in hypoxia condition, HIF-1α and VEGF can be induced significantly. These suggest serum starvation and hypoxia, two issues of ischemia, may activate HIF-1α and Shh signaling pathway individually in cardiomyoblast leading to cardiac protective mechanism in ischemia. We firstly demonstrated Shh induced by ischemia in cardiomyoblast and induced the downstream target genes expression. It is known that cardiomyocyte is a secretory cell and cardiac muscle is the major source of VEGF in the heart [22]. Here we show Shh, VEGF, and IGF-2 as paracrine or autocrine effectors are produced by cardiomyoblast to protect heart function.
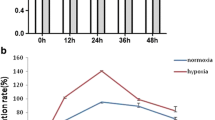
IGF-1 and IGF-2 were found to increase the proportion of embryos which formed blastocysts, and IGFs as mitogens and/or apoptotic survival factors during early bovine development. Perturbation of IGF-2 regulated growth may be involved in fetal oversize [23]. Hypoxia-induced increase in IGF-2 but not IGF-1 in primary osteoblasts [24]. In Sabra rat retina hypoxia model, retinal IGF-1 mRNA was reduced, as well as serum growth hormone (GH) [25]. In addition, after ischemia injury in brain, IGF-1 was progressed from vessels to tissues [26]. These suggest ischemia stimulated vessels permeability increase, to improve IGF-1 moving to tissues. Accordingly, in our study, IGF-1 and IGF-1R expressed aggregated in heart. However the mRNA and protein products were unchanged in cardiomyoblast cells after ischemia treatment. We suppose IGF-1 was secreted by some other mesenchymal cells in heart, or via endocrine circulating to heart to improve HIF-1α, Shh, and VEGF function and promote cell survival. Additionally, even though the growth factor IGF-2 had been induced in hypoxia, there was no proliferation effect on cardiomyoblast compared to normoxia condition (Fig. 5). We speculate that the hypoxia-induced damage was equal to repair maintaining cell viability in a short-term hypoxia, while IGF2 exerts a surviving rather than proliferating factor. Further more, once HIF-1α and Shh were inhibited, the cells viability was significantly decreased in hypoxia condition that firmly establish the importance of HIF-1α and Shh signals and their survival role leading to cardioprotective effect in hypoxia/ischemia attack (Fig. 6).

Inhibition of HIF-1α and Shh decrease cardiomyoblasts proliferation. Normoxic or hypoxic (24 h) cells in the presence and absence of 2ME2 (5, 10 μM) or cyclopamine (10, 20 μM) were assessed for MTT assay (optical absorption in 570 nm). Data showed proliferating inhibition in cells treated with inhibitors in both normoxia and hypoxia conditions. It means Shh and HIF-1α indeed govern proliferation effect of cardiomyoblast and there was a slightly increase in hypoxia versus normoxia condition, but was not significantly different. Data are triple replicated in independent experiments. (*P < 0.01, **P < 0.001, statistically different from hypoxic control; † P < 0.05, statistically different from normoxic control)

A depicted flow chart of ischemia/reperfusion induced HIF and Shh signaling may lead to angiogenesis and proliferation in cardiomyoblast cells by activating VEGF and IGF-2
The development of functional neovasculature in regenerating tissues requires precise spatial–temporal regulation of cell proliferation, migration, interaction, and differentiation. The role of Shh as a morphogen may be relevant to its potential activity to orchestrate appropriate postnatal angiogenesis after tissue injury [17]. However, the mechanism by which ischemia and/or hypoxia upregulate Shh expression remains uncertain. The promoter regions of Hh family members are not known to include a hypoxia-inducible factor sequence, and no data are available about the possible interactions between Shh- and hypoxia-inducible factor pathways in regulating VEGF and IGF-2 synthesis and stabilization. In this study, exposure of cells to inhibitor and downregulation of Shh and HIF-1α indicates the interaction between Shh and HIF-1α pathways.
Stimulated therapeutic angiogenesis to treat ischemic heart disease has been shown to be feasible in animal studies and is being tested in clinical trials [27–29]. The data we present here demonstrate the importance of Shh as a key regulator of angiogenesis when fetal heart under umbilical cord compression, and lend support to figure out ischemia injury induced signaling leading to protective effects to the fetal myocardium. Such considerations could have significant implications regarding the route of therapeutic administration of Shh.
References
Chi NC, Karliner JS (2004) Molecular determinants of responses to myocardial ischemia/reperfusion injury: focus on hypoxia-inducible and heat shock factors. Cardiovasc Res 61:437–447
Clark KE, Irion GL (1992) Fetal hemodynamic response to maternal intravenous nicotine administration. Am J Obstet Gynecol 167:1624–1631
Lyon HM, Holmes LB, Huang T (2003) Multiple congenital anomalies associated with in utero exposure of phenytoin: possible hypoxic ischemic mechanism? Birth Defects Res A Clin Mol Teratol 67:993–996
Carmeliet P, Dor Y, Herbert JM et al (1998) Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394:485–490
Feldser D, Agani F, Iyer NV et al (1999) Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res 59:3915–3918
Iyer NV, Kotch LE, Agani F et al (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12:149–162
Zhong H, Agani F, Baccala AA et al (1998) Increased expression of hypoxia-inducible factor 1α in rat and human prostate cancer. Cancer Res 58:5280–5284
Stiehl DP, Jelkmann W, Wenger RH et al (2002) Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1 beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett 512:157–162
Zelzer E, Levy Y, Kahana C et al (1998) Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO J 17:5085–5094
Zhong H, Chiles K, Feldser D et al (2000) Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545
Jetten AM (1991) Growth and differentiation factors in tracheobronchial epithelium. Am J Physiol 260:L361–L373
D’Ercole AJ (1987) Somatomedin/insulin-like growth factors and fetal growth. J Dev Physiol 9:481–495
Kobayashi S, Clemmons DR, Venkatachalam MA (1991) Colocalization of insulin-like growth factor binding protein with insulin-like growth factor-I. Am J Physiol 261:F22–F28
Grothey A, Voigt W, Schober C et al (1999) The role of insulin-like growth factor I and its receptor in cell growth, transformation, apoptosis, and chemoresistance in solid tumors. J Cancer Res Clin Oncol 125:166–173
Rappolee DA, Strum KS, Behrendsten O et al (1992) Insulin-like growth factor II acts through an endogenous growth pathway regulated by imprinting in early mouse embryos. Genes Dev 6:939–952
Grant MB, Mames RN, Fitzgerald C et al (1993) Insulin-like growth factor as an angiogenic agent: in vivo and in vitro studies. Ann NY Acad Sci 692:230–242
Pola R, Ling LE, Aprahamian TR et al (2003) Postnatal recapitulation of embryonic hedgehog pathway in response to skeletal muscle ischemia. Circulation 108:479–485
Pola R, Ling LE, Silver M et al (2001) The morphogen sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factor. Nat Med 7:706–711
Magal E, Goldin E, Harel S et al (1988) Acute uteroplacental ischemic embryo: lactic acid accumulation and prostaglandin production in the fetal rat brain. J Neurochem 51:75–80
Levy R, Glozman S, Milman D et al (2002) Ischemic reperfusion brain injury in fetal transgenic mice with elevated levels of copper-zinc superoxide dismutase. J Perinat Med 30:158–165
Nishimaki H, Kasai K, Kozaki KI et al (2004) A role of activated Sonic hedgehog signaling for the cellular proliferation of oral squamous cell carcinoma cell line. Biochem Biophys Res Commun 314:313–320
Giordano FJ, Gerber HP, Williams, SP et al (2001) A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA 98:5780–5785
Byrne AT, Southgate J, Brison DR et al (2002) Regulation of apoptosis in the bovine blastocyst by insulin and the insulin-like growth factor (IGF) superfamily. Mol Reprod Dev 62:489–495
Steinbrech DS, Mehrara BJ, Saadeh PB et al (2000) Hypoxia increases insulin-like growth factor gene expression in rat osteoblasts. Ann Plast Surg 44:529–535
Averbukh E, Weiss O, Halpert M et al (1998) Gene expression of insulin-like growth factor-1, its receptor and binding proteins in retina under hypoxic conditions. Metabolism 47:1331–1336
Guan J, Skinner SJ, Beilharz EJ et al (1996) The movement of IGF-1 into the brain parenchyma after hypoxic-ischemic injury. Neuroreport 7:632–636
Giordano FJ, Ping P, McKirnan MD et al (1996) Intracoronary gene transfer of fibroblast growth factor-5 increases blood flow and contractile function in an ischemic region of the heart. Nat Med 2:534–539
Hendel RC, Henry TD, Rocha-Singh K et al (2000) Effect of intracoronary recombinant human vascular endothelial growth factor on myocardial perfusion: evidence for a dose-dependent effect. Circulation 101:118–121
Laham RJ, Sellke FW, Edelman ER et al (1999) Local perivascular delivery of basic fibroblast growth factor in patients undergoing coronary bypass surgery: results of a phase I randomized, double-blind, placebo-controlled trial. Circulation 100:1865–1871
Acknowledgments
This work was supported by grant from the National Science Council of Republic of China (NSC94-2316-B-039-002).
Author information
Authors and Affiliations
Corresponding author
Additional information
Pei-Cheng Lin, Chih-Yang Huang, and Wei-Wen Kuo contributed equally to this work.
Rights and permissions
About this article
Cite this article
Hwang, JM., Weng, YJ., Lin, J.A. et al. Hypoxia-induced compensatory effect as related to Shh and HIF-1α in ischemia embryo rat heart. Mol Cell Biochem 311, 179–187 (2008). https://doi.org/10.1007/s11010-008-9708-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9708-6