
Abstract
An element/compound that acts as an antioxidant as well as, can increase the oxidative stress offers a new approach in differentiation therapy. Experiments were carried out to determine the effect of selenite on DNA damage and glutathione peroxidase (GPx) activity in N-nitrosodiethylamine (DEN) induced, phenobarbital promoted rat hepatoma. Supra-nutritional level of selenite (4 ppm) was supplemented at either, before-initiation/after-initiation and/or during entire period of the study. At the end of experiment period (20 weeks), extent of DNA damage (alkaline comet assay), selenium concentration, and GPx activity were assessed on nodular tissue (NL) cells, surrounding liver (SL) cells, and whole liver tissue (control) cells. Hepatic selenium level and GPx activity were decreased in DEN and PB-administered animals, whereas the DNA damage was found to be increased in both NL and SL cells compared with control group. However, the DNA damage is more in SL cells than in NL cells. Pre-supplementation of selenite did not show any difference in DNA (strand breaks) damage, selenium, and GPx activity. Increased hepatic selenium concentration and GPx activity were observed in both NL and SL cells in post-supplementation and entire period of selenite supplemented animals compared to DEN + PB treated animals. However, DNA damage was increased in NL but decreased in SL cells. Supplementation of selenite alone for 16 or 20 weeks had shown increased DNA damage, selenium concentration, and GPx activity compared to normal control animals. In summary, cancer bearing animals increased DNA damage and decreased Se level and GPx activity in NL and SL cells and other organs in cancer bearing animals, supplementation of Se further provoked DNA damage (no change in pretreatment) in NL cells, however it decreased DNA damage SL cells and other organs (kidney, lungs, and spleen). On the other hand Se levels and GPx activity were increased in NL and SL cells and other organs of Se-supplemented rats (no difference in group 3 animals). These results demonstrate that, in addition to chemopreventive and chemotherapeutic role of selenite, it also prevents cellular DNA damage induced in cancerous condition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The primary goal in the prevention of cancer and other mutation-related diseases is the avoidance of exposure to recognized risk factors. Strengthening of the host defense mechanisms provides a complementary preventive approach, which is particularly important when targeted to high-risk individuals. This strategy, referred to as chemoprevention, has found broad applications for the control of risk factors in cardiovascular diseases, and deserves greater emphasis in the prevention of cancer [1, 2]. The intake of protective factors can be achieved by means of both dietary measures and pharmacological agents.
The anticarcinogenic effect of selenium (Se) against various set of cancer-causing agents, including irradiation and carcinogens that form DNA adducts has been demonstrated in most organs examined in animal models [3, 4]. Several studies carried out in humans show an inverse correlation between Se intake and cancer incidence at several sites, including prostate, colon, lung, and breast [5–8]. Although the precise mechanism of Se anticancer activity remains to be determined, it is widely believed that multiple pathways are involved. It is at various stages of clinical development as a chemopreventive agent, demonstrating its ability to induce specific molecular perturbation associated with apoptosis and angiogenesis [9–12], by increasing phosphorylation of p53 mitogen-activated protein kinase, dephosphorylation of Akt and extracellular signal-regulated kinase 1/2, and PARP cleavage [9, 13, 14].
Various organic and inorganic Se compounds, generally considered to be antioxidants, produced mixed results when tested in animal models and human subjects. Among them, sodium selenite has been shown to be most effective in both in vitro and in vivo [4, 15–17]. Previous studies from our group have shown that sodium selenite treatment increases the overall antioxidant capacity of the hepatoma bearing animals [18, 19]. Recent studies demonstrate selenite, not only as an antioxidant, but possess oxidizing properties in the presence of specific substrates [20–22]. Nutritional essentiality of Se linked to the functional activities of several enzymes and proteins that contain Se, known as selenoproteins [21]. Several of these selenoproteins have antioxidant activities. Although the functions of most have not been determined, the effect of Se modulating the activity of these proteins could explain the possible mechanism by which Se might suppress the carcinogenesis. In the present investigation, we attempted to elucidate the possible role of sodium selenite as an in vivo antioxidant and/or oxidizing agent in chemical carcinogenesis.
In general, the indices used to measure DNA damage can be categorized into two subgroups. The first subgroup includes (i) single cell gel electrophoresis (the comet assay) [23] and (ii) terminal uridine nick end-labeling assay (TUNEL) [24]. The major advantages of these methods are that they directly measure/quantify DNA, are simple to perform, and focus on a single cell [25]. The second group comprising various biomarkers of DNA damage and repair, either in blood or urine samples, which indirectly measures/quantifies DNA damage and repair [26, 27]. Different reactive species react with different nucleic acid bases, for example, hydroxyl radicals react with all four nucleic acid bases whereas singlet oxygen reacts mainly with guanine [28].
In the present investigation, we employed Comet assay, which detects various forms of DNA strand breakage dependent on the pH of electrophoresis [29]. Under alkaline conditions (pH > 13), it detects single-strand breakage, double-strand breakage, excision repair site, and alkaline-labile sites [30]. Under neutral conditions, it mainly detects double-strand DNA breakage [31] and is therefore considered to be suitable for detection of DNA damage. The advantages of the comet assay for the detection of DNA damage are as follows: (a) it has higher sensitivity than the ladder assay [32] and TUNEL staining [33]; and (b) it can provide more specific information about the extent and heterogenity of DNA damage compared to TUNEL staining [34]. Based on hepatocarcinogeneic property and presence of various foods, we have chosen DEN as hepatocarcinogen initiator [4, 18, 19]. The purpose of the present study is essentially to determine the action(s) of selenite, an essential trace element that has shown a substantial inhibition on DEN-induced rat liver carcinogenesis. This may help us to further understand the inhibitory effect of selenite on the biochemical and biological aspects of DEN-induced and PB-promoted rat liver carcinogenesis.
Materials and methods
Animals and diet
Male, Wistar strain of albino rats, of age 6 weeks was used in these experiments. The rats were procured from Tamil Nadu Veterinary College, Chennai, India. They were fed with normal rat chow marketed by M/s.Hindustan Lever Limited, Mumbai, India and were provided with clean drinking water ad libitum. It was found that the rat chow used to feed our experimental animals contains 0.1 ppm of selenium, which is believed to satisfy the normal requirement of rats [35].
Chemicals and their sources
The following chemicals were purchased from the indicated sources: DEN, PB, bovine serum albumin, and sodium selenite from Sigma Chemical Co., (St. Louis, MO, USA). All other chemicals, including solvents, used were of high purity and analytical grade marketed by SD fine chemicals, Mumbai and Sisco Research Laboratories Pvt. Ltd., Mumbai, India.
Experimental design
The rats were divided into eight groups consisting of six animals in each (to study the mortality of experimental animals more numbers of rats were used). Liver tumors were induced in groups 2, 3, 5, and 7 with a single intraperitoneal injection of DEN at a dose of 200 mg kg−1 body weight in saline at the age of 10 weeks. Two weeks after DEN administration, the carcinogenic effect was promoted by the promoter, phenobarbital. Promoter was supplemented to the experimental animals through rat chow upto 14 successive weeks [18, 19].
Group 1 control animals were given the normal rat chow without additional selenite supplementation but the diet of groups 3, 4, 5, 6, 7, and 8 animals were supplemented with 4 ppm of selenium (as sodium selenite) in drinking water for various time periods as indicated below. Fresh drinking water supplemented with selenite was replaced on alternate days. The time point of DEN administration was taken as 0 (zero); minus (−) and plus (+) signs represents the time in weeks before and after DEN administration respectively. The schedule of selenium treatment in groups 3, 4, 5, 6, 7, and 8 was as follows: groups 3 and 4, −4 to 0; groups 5 and 6, +2 to +16; groups 7 and 8, −4 to +16. Groups, 4, 6, and 8 acted as selenium controls for groups 3, 5, and 7. The experiments were terminated 16 weeks after DEN administration (Fig. 1).

Schematic representation of experimental regimen
Twenty weeks after the initiation of the experiment, all the experimental animals were fasted overnight and killed by cervical decapitation. Liver was perfused in situ with 0.15 M NaCl at 37°C. Blood was collected and serum was separated. Hyperplastic nodules and non-nodular surrounding liver tissues were obtained from all the groups treated with DEN. The greyish-white hyperplastic nodules were easily identified from the surrounding reddish-brown liver tissues. Tissue samples from spleen, kidney, and lung were also collected for analyses.
Comet assays
Comet assay was performed by the method of Dhawan et al. [36] with slight modifications [37]. Lysis solution (without sodium sarcosinate and with 10% DMSO—freshly prepared), Tris–HCl neutralization (0.4 M, pH 7.5) buffer and electrophoresis (EP) buffer (300 mM NaOH, 1 mM EDTA) were prepared as described by Singh et al. [37]. In brief, the tissues were sliced with fine scissors on ice with ice-cold PBS, and cells (1 × 104) were suspended in 110 μl of low melting point agarose (0.65% LMPA-w/v in PBS, pH 7.4) and pipetted onto a frosted glass microscope slide precoated with 140 μl of 1% normal melting point agarose (NMPA) (in PBS, pH 7.4). The agarose was allowed to set for 10 min at 4°C and thereafter, the cover slip was removed and the slides were exposed for 24 h to lysis solution. Finally, the slides were rinsed with distilled water and EP buffer to remove salts. These slides were exposed to alkaline EP buffer (pH 13.0) for 40 min, and subjected to EP for 20 min (300 mA, 25 mV). Then the alkali was neutralized with Tris–HCl buffer; the slides rinsed with distilled water and methanol, and were stained with ethidium bromide.
Slide scoring
Slides were scored using nebug, an image analysis system attached to a fluorescence microscope equipped with appropriate filters. The microscope was connected to a computer through a charge coupled device (CCD) camera to transport images to software for analysis. The final magnification was ×400, the parameters taken for the liver cells were: tail DNA (%), tail length (migration of the DNA away from the nucleus, μm), and tail moment (arbitrary units). Images from 100 cells (50 each replicate slide/10 randomly selected different field) were analyzed.
Biochemical investigations
Se concentration was determined by the fluorometric method of Olson et al. [38]. The activity of glutathione peroxidase (GPx) was determined using hydrogen peroxide as substrate in the presence of reduced glutathione; estimation was carried out according to the method of Rotruck et al. [39]. The liver injury marker enzymes such as aspartate aminotransferase, lactate dehydrogenase, and γ -glutamyl transpeptidase were measured according to method of King [40, 41], and Massey and Williams [42] respectively. Total protein and albumin was estimated by the method of Reinhold [43]. For some biochemical assays, nodular or surrounding tissues were pooled together from different animals of the same group to get enough amounts of tissues.
Statistical analysis
Statistically significant (P < 0.05) differences between different groups were done using ANOVA and Student’s t-test. Each value in the results section represents two-way significance tests, i.e., b, represents significance against group 2 DEN-control and a, c, d, and e represents the same against their respective controls (groups 1, 4, 6, and 8).
Results
Food and water intake
During our experimental period, no differences in food and water consumption were observed between the different groups of animals. Food and water intakes were 11.5–14.6 g of diet/day/100 g of body weight and 8.5–11.5 ml of water/day/rat, respectively. A total of two rats from group 2 (16.6%) died before the end of the study. None of the rats from any other group died during the experimental period.
Changes in body weight and weights of organ
Table 1 shows the final body weight, liver, spleen, lung, and kidney of different groups of rats that were killed after 20 weeks of the study. The final body weight of DEN control rats (group 2) was significantly less (P< 0.01), where as liver weight is increased than that of the normal vehicle control (group 1). Supplementation of 4.0 ppm selenium for 20 consecutive weeks maintained the body weight at normal level and there were no significant differences between group 1 (normal vehicle control) and group 8 (selenium control) suggesting that selenium supplementation in this study did not have any adverse effect on the growth responses of the rats. Treatment with selenium for 20 weeks significantly increased (P< 0.05) the final body weight and reduced the liver weight of group 7 rats compared to the carcinogen control (group 2). There was no significant difference among the groups in their liver, spleen, lung, and kidney weights.
Se and GPx activity
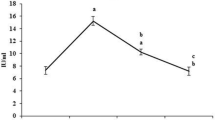
Table 2 shows the hepatic concentration of Se and GPx activity. The Se level and GPx activity are significantly decreased in NL and SL tissue of carcinogen bearing animals (group 2) when compared to group 1 animals. Pre-supplementation (group 3) of Se did not show any difference in Se levels and GPx activity when compared to group 2 animals. On the other hand post-supplementation and throughout the study, Se-treated groups (group 5 and 7) show increased level of Se and GPx activity. A significant difference in both Se and GPx activity was observed in both NL and SL tissues (except group 3) as compared with the values of the carcinogen (group 2) control rats and also with their respective controls, viz. groups 6 and 8.
Se level and GPx activity in lung, kidney, and spleen of different experimental groups are shown in Figs. 2 and 3. In the carcinogen control group (group 2), the GPx activity and Se level were found to be lower when compared with normal control animals (group 1). Se level and GPx activity are liable towards the normal value upon Se supplementation.

Levels of selenium in different experimental groups (details see Materials and methods). Each value represents mean ± SEM (n = 6); “a” as compared with group 1; “b” as compared with group 2; “c” as compared with group 4; “d” as compared with group 6; “e” as compared with group 8; (* P < 0.05, @ P < 0.01, # P < 0.001, NSnot statistically significant)

GPx activity in control and experimental animals. Groups were treated as mentioned in section Materials and methods. GPx activity is expresses as unit, one unit corresponds to μg of glutathione utilized/min/mg protein at 37°C, each value represents mean ± SEM (n = 6); “a” as compared with group 1; “b” as compared with group 2; “c” as compared with group 4; “d” as compared with group 6; “e” as compared with group 8; (* P < 0.05, @ P < 0.01, # P < 0.001, NSnot statistically significant)
Liver function test
The synthesizing capacity of the liver was reduced in carcinogen + promoter alone (group 2) treated rats, as indicated by decreased serum albumin, which was normalized by Se treatment for 20 weeks (group 7; Fig. 4a and b). Se treatment before initiation (group 3) and during promotion (group 5) also increased liver function 16.9% and 28.7% respectively. Liver injury, as estimated by serum LDH, AST, and α-GT, also improved significantly in group3 (∼18.29%), group 5 (∼34.38%), and group 7 (∼68.2%) Se-treated rats.

Level of albumin and ALT activity (a) and activities of LDH and γ-GT (b) in serum of control and experimental groups. Albumin level was expressed as unit, one unit corresponds to mg/dl of serum. ALT, LDH and γ-GT activities are expressed as units, for ALT and LDH one unit corresponds to μmoles of pyruvate liberated/minute/mg of protein at 37°C, for γ-GT one unit corresponds to nmoles of p-nitroaniline formed/minute/mg of protein at 37°C. Groups were treated as mentioned in section Materials and methods. Each value represents mean ± SEM (n = 6); “a” as compared with group 1; “b” as compared with group 2; “c” as compared with group 4; “d” as compared with group 6; “e” as compared with group 8; “f” as compared with group 7; (* P < 0.05, @ P < 0.01, # P < 0.001, NSnot statistically significant)
DNA damage
Figure 5 shows the DNA damage such as tail length (Fig. 6a), tail moment (Fig. 6b), % tail DNA (Fig. 6c) in control, and experimental animals. The carcinogen + promoter administered animals (group 2) the DNA damage is increased in NL (P < 0.001) and SL cells (P < 0.001) when compared with normal control group (group 1). SL cells showed more DNA (20%) damage than NL, if comparison made within group (group 2). Se supplementation for 4 weeks alone (before initiation of cancer-group3) did not show any statistical difference in DNA damage compared with carcinogen-treated group (group 2). However, the chemopreventive effect is not ruled out as reported by us earlier [18] and as shown in the present study. When Se was supplemented during promotion period (group 5), the DNA damage is increased in NL cells (P < 0.001), where as SL cells decreased (P < 0.01) when compared to group 2 animals. Group 6 animals compared with group 1 animals DNA damage is increased (P < 0.05). Se supplemented for 20 weeks, shown (group 7) significantly increased (P < 0.001) DNA damage in NL cells, whereas SL cells DNA damage was reduced (P < 0.001) when compared with group 2 animals. We did not observe any change (such as DNA laddering etc.) in DNA on agar gel electrophoresis and caspase-3 like activity (data not shown). This shows that there is no apoptotic-mediated cell death up on Se administration.

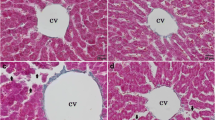
Effect of sodium selenite on DNA damage in control and experimental groups. Control (a) and selenite alone treated animals (d) for 20 weeks whole liver cells were used, carcinoma-bearing animals (b) and selenite (20 weeks) + carcinoma-bearing animals (c) nodular tissue cells were used. Thin arrow head shows non-DNA damaged cells; thick arrow head shows DNA damaged cells

Effect of sodium selenite on tail length (a), tail moment (b) and percent tail DNA (c) of whole liver tissue cells, nodular tissue (NL) and surrounding liver tissue (SL) cells of control and experimental animals. Groups were treated as mentioned in section Materials and methods. Each value represents mean ± SEM (n = 6); “a” as compared with group 1; “b” as compared with group 2; “c” as compared with group 4; “d” as compared with group 6; “e” as compared with group 8; “f” compared with group 2 NL Vs group 2 SL; (* P < 0.05, @ P < 0.01, # P < 0.001, NSnot statistically significant)
We measured DNA damage in lung, kidney, and spleen of control and experimental groups (Tables 3 and 4). In cancer bearing animals the DNA damage was increased considerably in lung, kidney, and spleen (P < 0.05; P < 0.01; P < 0.01, respectively). Se supplementation either before initiation alone (group 3) or during promotion alone (group 5) had no effect on these organs. Whereas Se supplementation for 20 weeks (group 7) reduced their DNA damage in lung (P < 0.05), kidney (P < 0.01) and spleen (P < 0.01). When pair fed control animals (group 4, 6, and 8) compare with normal control animals (group 1), there is no change in group 4 animals; but group 6 showed an increase in DNA damage in kidney and spleen, and group 8 showed an increase in kidney, spleen, and lung (all at P < 0.05).
Discussion
Previous studies from our laboratory demonstrated the chemopreventive and chemotherapeutic role of selenite, by increasing the oxidative defense molecules; which may be one of the mechanisms observed in multi-stage carcinogenesis [18, 19]. To further elucidate the possible in vivo mechanism of sodium selenite in hepatocarcinogenesis, we performed the present experiments. The extent of DNA damage (by comet assay) was measured as an indicator of oxidative stress. In addition, selenium and selenium-associated enzyme such as GPx was measured in the nodular, surrounding, and normal liver tissues of control and experimental groups. These parameters were employed based on the nature of anticancer drugs like cisplatin and mitomycin C, which are known to induce DNA damage in cancerous tissues; on the other hand antioxidant compounds such as quercetin and curcumin, which are also known for the anticancer effect through their antioxidant properties. However, to date as far as selenite is concerned, varying and conflicting results were reported [17, 44–46]. Hence, to understand the possible mechanism of action of selenite in vivo, we supplemented selenite either before initiation (group 3), during promotion (group 5), and through entire period of this study (group 7).
We observed reduction in the tissue selenium level, GPx activity, and increase in DNA damage in cancer-bearing animals not supplemented with Se. The increased DNA damage observed in cancer cells might be due to the reduced antioxidant capacity observed in cancerous animals [18, 19] or alteration in mineral content in cancer animals [47], for example: elevations in hepatic level of Fe2+, iron, though vital in life-processes, is also potentially toxic to living cells due to its ability to exist in two stable and inter-convertible redox-active states, since redox reactions catalyze the formation of oxyradicals generating superoxide radicals (O −2 ), which is the precursor of toxic H2O2 [48]. Moreover, ferrous iron can reduce copper to the cuprous state, which is a more potent generator of hydroxyl radicals than ferrous ions, thus iron acts synergistically with copper in the carcinogenic process. Thus, antioxidant-deficient environment and accumulated free radicals ultimately favor DNA lesions resulting in increased hepatic cell proliferation, phenotypic transformation, and expression of neoplastic pathology with minimal apoptotic events. Se-mediated restoration of hepatic levels of antioxidant [18] may have a role in the repair of DNA base-lesions in vivo. Furthermore, normalization of hepatic Fe levels [48] minimizes the possibility of free-radical generation, thereby preventing oxidative injury to cells and DNA. Restoration of antioxidant level after treatment with Se has been linked with suppression of cell proliferation events [49]. Studies from our laboratory indicate that, at a dose of 4.0 ppm increases the antioxidant levels and maintains membrane integrity [18, 50].
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated as a consequence of carcinogen exposure, leading to DNA-strand breaks [51]. For the cell, double-strand breaks (DSBs) are probably the most deleterious form of DNA damage and may arise during the replication of single strand breaks (SSBs), when carcinogen-induced SSBs remain unrepaired [52]. Error-prone repair of DSBs can lead to chromosomal aberrations as well as oncogene activation, which contribute to carcinogenesis. Thus, SSBs can be considered as a fundamental to the maintenance of chromosome integrity and genetic stability. In the present study, a substantial decrease in the amount of DEN-induced ‘tailed’ DNA and DNA ‘comets’ by Se could reflect its anticlastogenic potential to combat genotoxicity. Selenite reacts with reduced form of glutathione (GSH) in the metabolic process leads to the formation of ROS, (see below).

Another potential mechanism of Se that comes out from our study is Se-mediated induction of DNA damage particularly in cancer cells. The involvement of free radical in many degenerating diseases including cancer based on the detection of the oxidation products of nucleic acids, proteins, and lipids formed as a consequence of diseased condition. Tumor growth is associated with tissue hypoxia that is accompanied by the formation of reductive rather than oxidative free radicals. Although, the most biologically active oxidant such as hydroxyl radical has been generated during hypoxia [53, 54]. Possible mechanism by which selenite causes more DNA damage in cancerous cells compared to surrounding cells is that cancer cell membrane-bound proteins are associated with polythiols, which appear under the reducing conditions of hypoxic tumor cells. These thiol groups can, in turn, initiate a disulfide exchange reaction with plasma proteins, predominantly with fibrinogen, to form an insoluble and protease-resistant fibrin-like polymer. As the result, tumor cells become surrounded by a coat that masks specific tumor antigens thus allowing cancer cells to escape immune recognition and elimination by natural killer cells [55–57]. Selenite is capable of oxidizing polythiols to corresponding disulfides, but does not react with monothiols. Selenite by virtue of oxidizing cell membrane thiols, can prevent the formation of the coat and consequently makes cancer cells vulnerable to the immune surveillance and destruction [55, 56].
Reduced glutathione is involved in Se metabolism and its bioactivity. Previous study from our group found that cancer cells have low levels of reduced glutathione [19]. Study by Shen et al. [25] shows that both increase and depletion of reduced glutathione content enhances the selenite-induced oxidative stress and apoptosis in human hepatoma cells. It should be pointed out, that there are two fundamental differences between the group 5 versus group 3. Pretreatment of selenite for 4 weeks (group 3), inhibited tumor incidence around ∼25% where as group 5 (16 weeks of post-treated selenite) shows only ∼15–18% inhibition [4]. However, there is no difference in the extent of DNA damage in group 3 but group 5 showed increased DNA damage in cancer cells. Selenite post-treated group showed further increase in DNA damage with minimal antitumor activity, these results shows that sodium selenite play a dual role in chemical carcinogenesis. For example, short time supplementations of Se in normal animals increase the intracellular GSH levels [58, 59]. In vitro studies show controversial reports on the changes in intracellular GSH [60–62]. We believe, short-time supplementation of selenite might increase some of the selenium-related proteins and/or GSH [58, 59]. Increased GSH may reduce the carcinogen-DNA interaction or increase carcinogen metabolism, which in turn reduce tumor incidence. Continuous supplementation of Se may result in the generation of oxidative stress, which has been proposed as one of the mechanisms by which this element exerts its cellular actions in cancer cells.
Selenium potentially affect cancer development through its oxidative stress, DNA repair, inflammation, apoptosis, proliferation, carcinogen metabolism, testosterone production, angiogenesis, fat metabolism, and immune function [13, 44, 63, 64]. Natural organic (e.g., selenomethionine) and inorganic (e.g., selenite) forms of Se are metabolized via different pathways into selenide, which then be either phosphorylated and ultimately incorporated as selenocysteine into active selenoproteins or methylated into active metabolites, such as methylselenol [65, 66]. Therefore, the effect of Se can be indirect (via incorporation into selenoproteins) and/or direct (via selenium metabolites). Direct effect of Se varies with different metabolites, in normal versus malignant prostate cells. The most active known metabolites in preclinical studies are natural methylated compounds (e.g., methylselenol) and synthetic organoselenium compounds (e.g., 1,4 phenylenebis(methylene) selenocyanate) [65, 66]. The molecular targets include manganese superoxide dismutase, p21, caspase-8, NF-κB, protein kinase C, and the androgen receptor in prostate cancer [17, 44, 67–71]. Selenium indirectly effects via enzymatic functions of certain selenoproteins. Besides their well-known effects (e.g., of glutathione peroxidase) on intracellular redox, selenoproteins posses other activities, which varies with cell type, physiologic status, presence or absence of incorporated selenocysteine. For example, selenoprotein thioredoxin reductase without (but not with) selenocysteine appears to induce apoptosis and inhibit growth in certain cell types [72, 73].
Results from our present study demonstrate that sodium selenite increases DNA damage, selenium level, and GPx activity in NL cells, whereas decreases the DNA damage in SL cells and other organs of cancer-bearing animals. We also found that long-term supplementation of Se (group 6 and 8) alone causes DNA damage. On the other hand, these animals did not show any harmful effect such as weight loss, reduced food intake and liver toxicity. To elucidate exact mechanism(s) that how Se increases DNA damage in NL cells and decreased in SL cells further studies are needed.
References
Chemoprevention Working Group (21 members) (1999) Prevention of cancer in the next millennium: report of the Chemoprevention Working Group to the American Association for Cancer Research. Cancer Res 59:4743–4758
De Flora S, Izzotti A, D’Agostini F, Balansky RM, Noonan D, Albini A (2001) Multiple points of intervention in the prevention of cancer and other mutation related diseases. Mutat Res 480–481:9–22
El-Bayoumy K (1991) The role of selenium in cancer prevention. In: DeVita VT, Hellman S, Rosenberg SA (eds) Cancer prevention. J. B. Lippincott, Philadelphia, PA, pp 1–15
Thirunavukkarasu C, Jagadeeswaran R, Babu E, Sakthisekaran D (2000) Inhibitory effect of selenium on N-nitrosodiethylamine and Phenobarbital—promoted rat liver carcinogenesis. J Clin Biochem Nutr 28:69–80
Yoshizawa K, Willett WC, Morris SJ, Stampfer MJ, Spiegelman D, Rimm EB, Giovannucci E (1998) Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst 90:1219–1224
Ghadirian P, Maisonneuve P, Perret C, Kennedy G, Boyle P, Krewski D Lacroix A (2000) A case-control study of toenail selenium and cancer of the breast, colon, and prostate. Cancer Detect Prev 24:305–313
Brooks JD, Metter EJ, Chan DW, Sokoll LJ, Landis P, Nelson WG, Muller D, Andres R, Carter HB (2001) Plasma selenium level before diagnosis and the risk of prostate cancer development. J Urol 166:2034–2038
Fernandez-Banares F, Cabre E, Esteve M, Mingorance MD, Abad-Lacruz A, Lachica M, Gil A, Gassull MA (2002) Serum selenium and risk of large size colorectal adenomas in a geographical area with a low selenium status. Am J Gastroenterol 97:2103–2108
Wang Z, Jiang C, Ganther H, Lu J (2001) Antimitogenic and proapoptotic activities of methylseleninic acid in vascular endothelial cells and associated effects on PI3K-AKT, ERK, JNK, and p38 MAPK Signaling. Cancer Res 61:7171–7178
Wang Z, Jiang C, Lu J (2002) Induction of caspase-mediated apoptosis and cell cycle G1 arrest by selenium metabolite methylselenol. Mol Carcinog 34:113–120
Gudkov A (2002) Converting p53 from a killer into a healer. Nat Med 8:1196–1198
Baines A, Taylor-Parker M, Goulet A, Renaud C, Gerner E, Nelson M (2002) Selenomethionine inhibits growth and suppresses cyclooxygenase-2 (COX-2) protein expression in human colon cancer cell lines. Cancer Biol Ther 4:370–374
Ip C, Dong Y (2001) Methylselenocysteine modulates proliferation and apoptosis biomarkers in premalignant lesions of the rat mammary gland. Anticancer Res 21:863–867
Lu J, Jiang C, Kaeck M, Ganther H, Vadhanavikit S, Ip C, Thompson H (1995) Dissociation of the genotoxic and growth inhibitory effects of selenium. Biochem. Pharmacol 50:213–221
Lipinski B (2005) Rationale for the treatment of cancer with sodium selenite. Med Hypotheses 64:806–810
Wycherly BJ, Moak MA, Christensen MJ (2004) High dietary intake of sodium selenite induces oxidative DNA damage in rat liver. Nutr Cancer 48:78–83
Al-Taie OH, Seufert J, Karvar S, Adolph C, Mork H, Scheurlen M, Kohrle J, Jakob F (2003) Selenium supplementation enhances low selenium levels and stimulates glutathione peroxidase activity in peripheral blood and distal colon mucosa in past and present carriers of colon adenomas. Nutr Cancer 46:125–130
Thirunavukkarasu C, Sakthisekaran D (2001) Effect of selenium on N-nitrosodiethylamine-induced multistage hepatocarcinogenesis with reference to lipid peroxidation and enzymic antioxidants. Cell Biochem Funct 19:27–35
Thirunavukkarasu C, Babu E, Ebrahim AS, Chandramohan N, Sakthisekaran D (2004) Antioxidant-associated chemoprevention by sodium selenite in N-nitrosodiethylamine-induced and phenobarbital-promoted hepatocarcinogenesis in rats. Cell Biochem Funct 22:265–271
Ip C (1998) Lessons from basic research in selenium and cancer prevention. J Nutr 128:1845–1854
Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN (2003) Characterization of mammalian selenoproteomes. Science 300:1439–1443
Raich PC, Lu J, Thompson HJ, Combs GF Jr (2001) Selenium in cancer prevention: clinical issues and implications. Cancer Invest 19:540–553
Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM (2001) Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 88:733–739
Kockx MM, Muhring J, Knaapen MW, de Meyer GR (1998) RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am J Pathol 152:885–888
Shen H, Ong C (2000) Detection of oxidative DNA damage in human sperm and its association with sperm function and male infertility. Free Radic Biol Med 28:529–536
Halliwell B (2000) Why and how should we measure oxidative DNA damage in nutritional studies? How far have we come? Am J Clin Nutr 72:1082–1087
Lindahl T, Wood RD (1999) Quality control by DNA repair. Science 286:1897–1905
Rehman A, Nourooz-Zadeh J, Moller W, Tritschler H, Pereira P, Halliwell B (1999) Increased oxidative damage to all DNA bases in patients with type II diabetes mellitus. FEBS Lett 448:120–122
Collins AR (2002) Comet assay—principles, applications, and limitations. Methods Mol Biol 203:163–177
Abt G, Vaghef H, Gebhart E, Dahlgren CV, Hellman B (1997) The role of N-acetylcysteine as a putative radioprotective agent on X-ray-induced DNA damage as evaluated by alkaline single-cell gel electrophoresis. Mutat Res 384:55–64
Olive PL, Wlodek D, Banath JP (1991) DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res 51:4671–4676
Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D (2002) DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation of H(2)O(2). Free Radic Biol Med 33:691–702
Godard T, Deslandes E, Lebailly P, Vigreux C, Sichel F, Poul JM, Gauduchon P (1999) Early detection of staurosporine-induced apoptosis by comet and annexin V assays. Histochem Cell Biol 112:155–161
Kindzelskii AL, Petty HR (2002) Ultrasensitive detection of DNA damage by the combination of the comet and TUNEL assays. Methods Mol Biol 203:195–201
Newberne PM, Bieri JG, Briggs GM, Nesheim MC (1978) Control of diets in laboratory animal experimentation. Inst Lab Anim Resou News 21:A1–A12
Dhawan A, Mathur N, Seth PK (2001) The effect of smoking and eating habit on DNA damage in Indian populations as measured in the comet assay. Mutat Res 474:121–128
Singh NP, McCoy MT, Tice RR, Schnider EL (1988) A simple technique for the quanitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184–191
Olson OE, Palmer SI, Carey EE (1975) Modification of the official fluorometric method for selenium in plants. J Assoc Off Anal Chem 58:117–121
Rotruck JT, Pope AL, Ganther EH, Swanson AB, Hafeman GD, Hoekstra GW (1973) Selenium biochemical role as a component of glutathione peroxidase. Science 179:588–590
King J (1965) The dehydrogenases or oxido reductase-lactate dehydrogenase. In: King J (ed) Practical clinical enzymology. D Van Nostrand Company Ltd., London, pp 83–93
King J (1965) The transferase—alanine and aspartate transaminase. In: King J (ed) Practical clinical enzymology. D Van Nostrand Company Ltd., London, pp 121–138
Massey V, Williams CH (1965) On the reaction mechanism of yeast glutathione reductase. J Biol Chem 240:4470–4480
Reinhold JG (1953) Manual determination of serum total protein albumin and globulin fractions by Buiret method. In: Reiner M (ed) Standard methods in clinical chemistry, vol 1. Academic Press, New York, p 88
Meuillet E, Stratton S, Cherukuri DP, Goulet AC, Kagey J, Porterfield B, Nelson MA (2004) Chemoprevention of prostate cancer with selenium: an update on current clinical trials and preclinical findings. J Cell Biochem 91:443–458
Leist M, Maurer S, Schultz M, Elsner A, Gawlik D, Brigelius-Flohe R (1999) Cytoprotection against lipid hydroperoxides correlates with increased glutathione peroxidase activities, but not selenium uptake from different selenocompounds. Biol Trace Elem Res 68:159–174
Thompson HJ, IP C (1991) Temporal changes in tissue glutathione in response to chemical form, dose, and duration of selenium treatment. Relevance to cancer chemoprevention by selenium. Biol Trace Elem Res 30:163–173
Thirunavukkarasu C, Sakthisekaran D (2003) Effect of dietary selenite on N-nitrosodiethylamine-induced and phenobarbital promoted multistage hepatocarcinogenesis in rat: reflection in some minerals. Biomed Pharmacother 57:416–421
Keyer K, Imlay A (1996) Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA 93:13635–13640
Zheng QS, Zheng RL (2002) Effects of ascorbic acid and sodium selenite on growth and redifferentiation in human hepatoma cells and its mechanisms. Pharmazie 57:265–269
Thirunavukkarasu C, Sakthisekaran D (2003) Stabilization of membrane bound enzyme profiles by sodium selenite in N-nitrosodiethylamine induced and phenobarbital promoted hepatocarcinogenesis in rats. Biomed Pharmacother 57:117–123
Halliwell B, Aruoma OI (1991) DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian cells. FEBS Lett 281:9–19
Jackson SP (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis 23:687–696
Samarasinghe DA, Tapner M, Farrell GC (2000) Role of oxidative stress in hypoxia-reoxygenation injury to cultured rat hepatic sinusoidal endothelial cells. Hepatology 31:160–165
Younes M, Kayser E, Strubelt O (1992) Effect of antioxidants on hypoxia/reoxygenation-induced injury in isolated perfused rat liver. Pharmacol Toxicol 71:278–283
Lipinski B (2005) Rationale for the treatment of cancer with sodium selenite. Med Hypotheses 64:806–810
Tarze A, Dauplais M, Grigoras I, Lazard M, Ha-Duong NT, Barbier F, Blanquet S, Plateau P (2007) Extracellular production of hydrogen selenide accounts for thiol-assisted toxicity of selenite against Saccharomyces cerevisiae. J Biol Chem 282:8759–8767
Drake EN (2006) Cancer chemoprevention: selenium as a prooxidant, not an antioxidant. Med Hypotheses 67:318–322
Chakrabarti S, Brodeur J (1985) Influence of selenium on the metabolism of bromobenzene and a possible relationship to its hepatotoxicity. Environ Res 37:327–339
Yin SA, Sato I, Hosokawa Y, Niizeki S, Tojo H, Yamaguchi K (1991) Effects of dietary zinc and cadmium on tissue selenium concentration and glutathione peroxidase activity in rats fed DL-selenomethionine or sodium selenite. J Nutr Sci Vitaminol 37:29–37
Bell RR, Nonavinakere VK, Soliman MR, Early JL (1991) Effect of in vitro treatment of rat hepatocytes with selenium, and/or cadmium on cell viability, glucose output, and cellular glutathione. Toxicology 69:111–119
Chung AS, Maines MD, Reynolds WA (1982) Inhibition of the enzymes of glutathione metabolism by mercuric chloride in the rat kidney: reversal by selenium. Biochem Pharmacol 31:3093–3100
Anundi I, Hogberg J, Stahl A (1982) Involvement of glutathione reductase in selenite metabolism and toxicity, studied in isolated rat hepatocytes. Arch Toxicol 50:113–123
Rayman MP (2000) The importance of selenium to human health. Lancet 356:233–241
Hatfield DL, Gladyshev VN (2002) How selenium has altered our understanding of the genetic code. Mol Cell Biol 22:3565–3576
Tapiero H, Townsend DM, Tew KD (2003) The antioxidant role of selenium and seleno-compounds. Biomed Pharmacother 57:134–144
Ip C, Thompson HJ, Zhu Z, Ganther HE (2000) In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res 60:2882–2886
Menter DG, Sabichi AL, Lippman SM (2000) Selenium effects on prostate cell growth. Cancer Epidemiol Biomarkers Prev 9:1171–1182
Zhong W, Oberley TD (2001) Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res 61:7071–7078
Jiang C, Wang Z, Ganther H, Lu J (2001) Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res 61:3062–3070
Venkateswaran V, Klotz LH, Fleshner NE (2002) Selenium modulation of cell proliferation and cell cycle biomarkers in human prostate carcinoma cell lines. Cancer Res 62:2540–2545
Dong Y, Lee SO, Zhang H, Marshall J, Gao AC, Ip C (2004) Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res 64:19–22
Gallegos A, Berggren M, Gasdaska JR, Powis G (1997) Mechanisms of the regulation of thioredoxin reductase activity in cancer cells by the chemopreventive agent selenium. Cancer Res 57:4965–4970
Anestal K, Arner ES (2003) Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocytsteine. J Biol Chem 278:15966–15972
Acknowledgment
One of the authors, Dr. C. Thirunavukkarasu, gratefully acknowledges the Council of Scientific and Industrial Research, New Delhi, India for the financial assistance in the form of Senior Research Fellowship [9/115(484)/99–EMR-I).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thirunavukkarasu, C., Premkumar, K., Sheriff, A.K. et al. Sodium selenite enhances glutathione peroxidase activity and DNA strand breaks in hepatoma induced by N-nitrosodiethylamine and promoted by phenobarbital. Mol Cell Biochem 310, 129–139 (2008). https://doi.org/10.1007/s11010-007-9673-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-007-9673-5