
Abstract
Most existing antimicrobial peptides (AMPs) are α-helical and cationic that exhibit typical amphipathic feature to facilitate efficient interaction with bacterial outer membranes. However, short α-helix is unstable in water, and thus naturally occurring α-helical AMPs are generally long and structurally complex. Here, we attempt to perform de novo design of very simple AMPs with only a single-turn α-helix by using hydrocarbon-stapling technique, which can effectively constrain peptide conformation into helical form. The designed AMPs are heptapeptides that have an additional residue at each end of pentapeptides, the theoretical minimum to define an α-turn. Net charge, amphipathicity, sequence pattern and amino acid composition are systematically considered and examined based on the helical wheel of standard α-helical heptapeptide, which derive a series of potential one-turn unstapled AMP candidates with strong hydrophobic moment and a good balance between cationicity and hydrophobicity. Structural analysis suggests that, however, these designed AMPs cannot spontaneously fold into α-helical conformation, with helicity < 50%. An all-hydrocarbon bridge is stapled across the 2nd and 6th residues of several selected heptapeptides to arbitrarily force their α-helical propensity. Circular dichroism spectroscopy demonstrates that the stapling can largely enhance the helical content of these heptapeptides. Molecular dynamics simulation reveals that, as compared to unstapled peptides, the stapled peptides can more efficiently penetrate into the membrane surface of a lipid bilayer model and swiftly move across the hydrophilic surface layer. Susceptibility test reveals that the stapling can considerably improve the antibacterial potency of designed peptides against both the antibiotic-sensitive and methicillin-resistant S. aureus; permeabilization assay confirms that the stapled peptides generally have a higher permeability on E. coil outer membrane than their unstapled counterparts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Peptide segments of 10–20 amino acids long with α-helical conformation were estimated by Barlow and Thornton to account for more than 30% of protein secondary structures (Barlow and Thornton 1988). They have been frequently observed to regulate diverse biological functions through peptide-mediated interactions with their partner proteins, DNA or RNA. For example, crystal structure survey suggested that roughly 62% of protein complexes constitute α-helical interfaces (Jochim and Arora 2010), which were identified as potential targets for synthetic modulators of protein–protein interactions (Jochim and Arora 2009). In addition, small helical peptide segments have been found as self-binding peptides, which serve as molecular switch to regulate the biological activity of their parent proteins (Yang et al. 2015a, 2016; Bai et al. 2017), and bioinformatics methods such as machine learning and molecular docking have been successfully used to minimize the functional structures of small helical peptides (Ren et al. 2011; Li et al. 2019a, b; Luo et al. 2015). However, short synthetic peptides corresponding to such helical motifs tend not to form appreciable helical structure in water, away from the helix-stabilizing hydrophobic environments of proteins (Scholtz and Baldwin 1992). If short peptide α-helices could be stabilized or mimicked in water by small molecules, such compounds might be valuable chemical or biological probes and lead to development of novel pharmaceuticals, vaccines, diagnostics, biopolymers, and industrial agents (Shepherd et al. 2005).
The α-helix is a common conformation adopted by many natural and synthetic antimicrobial peptides (AMPs), which can define the amphipathic helical motif that facilitates efficient interaction with bacterial outer membranes (Tossi et al. 2000). However, since short α-helix cannot stably exist in water, most known α-helical AMPs are longer than 15 amino acid residues, and only very few have shorter sequence length (e.g. 10–15 amino acids). Currently, the shortest α-helical AMPs deposited in the APD3 database (Wang et al. 2016) are natural octapeptide Temporin-SHf and synthetic octapeptide TetraF2W-RK. However, both the two peptides have been found to have a moderate content of structural disorder in water (Abbassi et al. 2010; Mishra et al. 2017), suggesting that they cannot be well stabilized in structured conformation and possess only a partial α-helix.
An α-turn, the minimal repeating unit in an α-helix, is defined by a closed ring formed by a hydrogen bond between a carbonyl oxygen (>C=O) of one residue (i) and the amide hydrogen (–NH) of another residue (i + 4) separated by three residues, suggesting that five residues is the theoretical minimum to define an α-turn (Shepherd et al. 2005). However, such short α-helix cannot be spontaneously formed without protein context support (Zhou et al. 2018, 2019). In order to stabilize the α-helical conformation of short peptides in water, all-hydrocarbon stapling technique was used in this study to examine the possibility of developing very small cationic AMPs with only single helical turn. Although five amino acids are enough to form a single turn of helix, it might be too short to effectively supply amphipathicity required for the antimicrobial activity of designed peptides. Therefore, we herein considered heptapeptides, in which an additional residue at each end of one helical turn would provide supplementary options to manipulate their physiochemical properties (Dinh et al. 2014, 2015; Luong et al. 2017). Hydrocarbon stapling has been widely used to constrain peptides into bioactive helical conformation (Walensky and Bird 2014), which has also been successfully applied to designing helical AMPs (Migoń et al. 2018). Here, we attempted to develop a standard modeling strategy that can be used in the de nono design of single-turn α-helical, cationic AMPs without any priori knowledge, such as use of known longer AMPs as structural start or template. With the strategy we successfully designed several (i, i + 4) hydrocarbon-stapled heptapeptides and demonstrated that the stapling can promote the folding of very short peptides to amphipathic α-helix and effectively stabilize the single-turn helical structure in water. The complete dynamics trajectory and conformational flexibility of peptide interaction with the outer membrane model of Gram-positive bacteria were also investigated in detail using dynamics simulation and conformational analysis. In addition, the antibacterial activity and solution conformation of stapled peptides and their unstapled counterparts were measured using susceptibility test, permeabilization assay and circular dichroism.
Materials and Methods
Susceptibility Test
The unstapled peptides with C-terminal amidation (–NH2) were synthesized using Fmoc solid phase chemistry. For hydrocarbon-stapled peptides, the Grubbs catalysts were used in olefin metathesis to incorporate the all-hydrocarbon staples into peptides (Bird et al. 2011). The prepared peptides were resuspended in water or phosphate-buffered saline (PBS) to a concentration of 10 mg/ml and stored at − 20 °C. The CLSI microbroth dilution susceptibility test described previously (Yau et al. 2001; Frecer et al. 2004) was used to determine the minimum inhibitory concentration (MIC) of peptides against two strains of Staphylococcus aureus, i.e. antibiotic-sensitive S. aureus (ATCC25923) and methicillin-resistant S. aureus (MRSA). The strains were cultured in 10 ml of Mueller–Hinton broth (MHB) at 37 °C and diluted to reach a final cell population of ~ 5 × 105 CFU/ml. To determine the effect of peptides on bacterial growth kinetics, 100 μl bacterial suspensions incubated with peptides at different concentrations were placed in a microplate reader overnight and the optical density was recorded. The minimum inhibitory concentration (MIC) was determined as the lowest concentration of peptide that reduced 50% bacterial growth. Each assay was performed in triplicate.
Circular Dichroism
Circular dichroism (CD) spectra of stapled and unstapled peptides in water solution were analyzed at room temperature by using a J-810 spectropolarimeter. The peptide samples were added to obtain a final concentration of 25 μM in 10 mM sodium phosphate buffer (pH 7.4). In addition, the CD spectra of stapled and unstapled p(3,7) peptides in 0% and 50% TFE solutions (TFE:H2O = 0:100 and 50:50, respectively) were also tested to compare the effects of water and lipid environments on peptide structures. The samples were loaded in a rectangular quartz cell with 0.1 cm path length, and the spectra were monitored at a 10 nm/min scanning speed in wavelength range 190–250 nm (Wang et al. 2019). The final spectra were expressed as mean residue ellipticity [θ] in deg cm2 dmol−1, the molar ellipticity per residue. The percentage of peptide helical content (%helicity) was calculated with spectral records at 222 nm (Greenfield 2006).
Membrane Permeabilization
The outer membrane permeability of stapled and unstapled peptides was determined by using N-phenyl-1-napthylamine (NPN) uptake assays with E. coli strain UB1005 (Lee et al. 2004; Lv et al. 2014). Briefly, the strain was suspended in HEPES buffer (pH 7.4) containing 5 mM glucose. 10 mM NPN was added to cells in a quartz cuvette, and the tested peptides with varying concentrations were then added to the cuvette, for which the fluorescence was recorded. The permeability of bacterial outer membrane can be increased by peptide disruption, which promotes NPN interaction with the membrane and thus results in an increased fluorescence. Consequently, the NPN uptake can be calculated as: uptake = 100% × (Fobs − Fo)/(FPB − Fo), where Fo is the background fluorescence of NPN without peptides, Fobs is the observed fluorescence of NPN with a particular peptide concentration, and FPB is the fluorescence of NPN with 10 μg/ml positive control (polymyxin b).
Dynamics Simulation
The start structures of stapled p(3,7) peptide and its unstapled counterpart were assigned with helical and linear conformations, respectively. The peptide interaction dynamics with bacterial outer membrane was investigated with atomistic molecular dynamics (MD) simulations in the GROMACS ver. 4.5.5 package (Van Der Spoel et al. 2005; Yu et al. 2014). The simulations were essentially similar to previous reports (Fu et al. 2018; Bai and Chen 2019). A solvated lipid bilayer model was set up with a composition of mixed lipids POPG:POPE (3:1) to mimic the zwitterionic outer membrane of Gram-positive bacteria (Yang et al. 2018; Ye 2018). The OPLS force field was used to describe stapled and unstapled peptides (Jorgensen et al. 1996), and the force field parameters for lipid models were taken from Kukol et al. (2009). Since the OPLS is an all-atom force field, it can be directly applied to hydrocarbon bridge. Previously, the force field has been successfully used to perform MD simulations of hydrocarbon-stapled peptides in the GROMACS environment (De Paola et al. 2015). The created lipid bilayer consisted of 72 lipid molecules and ~ 4000 TIP3P water molecules in each monolayer. The investigated peptide was placed ~ 40 Å away from the membrane center. Subsequently, 500-ns MD simulations with 2 fs time step were performed for each system under NPT ensemble (Yang et al. 2015b; Zhou et al. 2016), with water, lipids and the peptide coupled separately to a heat bath with T = 298 K and a time constant τ = 0.1 ps using weak temperature coupling, and atmospheric pressure was maintained at 1 bar using weak semi-isotropic pressure coupling (Wang et al. 2014). During the simulations particle-mesh-Ewald (PME) method was applied to long electrostatic interactions (Darden et al. 1993) and LINCS algorithm was used to treat covalent bonds involving hydrogen atoms (Hess et al. 1997).
Results and Discussion
De novo Design of Single-Turn Amphipathic α-Helical Heptapeptides
It is known that short peptides are hard to be stabilized in α-helical conformation without protein context support (Shepherd et al. 2005). By searching against the APD3 database (Wang et al. 2016), only very few short AMPs are assigned as helical structure, including two octapeptides, two nanopeptides and three decapeptides (Table 1). They are all cationic AMPs with + 1 to + 4 net charges. Most of these peptides have moderate or high hydrophobicity (H > 0.5) and hydrophobic moment (µ > 0.4). In addition, they were also predicted to considerably or partially fold into α-helical conformation, with helicity > 50%. However, there are two outliers, i.e. Cn-AMP1 and Cm-p1, which have relatively low helicity, hydrophobicity, hydrophobic moment (h = 37.9% and 28.2%; H = 0.200 and 0.189; µ = 0.265 and 0.057, respectively), suggesting that the two peptides may exert their antibacterial activity via a non-amphipathic helical mechanism. In this respect, we considered that a short amphipathic AMP should be helical (h > 50%) and cationic (c > + 1) as well as have high hydrophobicity (H > 0.5) and hydrophobic moment (µ > 0.4).
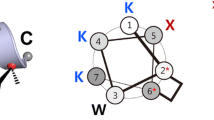
Although pentapeptide is the minimal length requirement for a single turn of α-helix, it might be too short to effectively supply amphipathicity required for the antimicrobial activity of designed peptides. Therefore, we considered heptapeptides, in which an additional residue at each end of one helical turn would provide supplementary options to manipulate their physiochemical properties (Dinh et al. 2014, 2015). Since the cationic AMPs should be positively charged but also have a high hydrophobicity, we considered a moderate net charge (+ 2 or + 3) for the peptides. In addition, according to a previous statistical survey the positively charged Lys (K) and Arg (R) as well as the hydrophobic/aromatic Leu (L) and Trp (W) contribute the highest amino acid composition abundance to α-helical AMPs (Mishra and Wang 2012). Therefore, we considered their four possible combinations to define an AMP, i.e. (K, L) (K, W) (R, L) and (R, W). The helical wheel of a standard one-turn α-helical heptapeptide is shown in Fig. 1, from which the residue distribution around the wheel is readily visualized, that is, in turn, X1 → X5 → X2 → X6 → X3 → X7 → X4. Here, 14 AMP sequence patterns were designed based on the residue distribution order of helical wheel, including 7 patterns with + 2 net charge and 7 patterns with + 3 net charge; they represent totally 56 heptapeptides (see Table S1 in Supplementary Materials). As can be seen, these designed peptides with different sequence patterns exhibit an appreciable hydrophobicity (H > 0.4) and a moderate or high hydrophobic moment (µ > 0.5) as predicted by HeliQuest server (Gautier et al. 2008). The + 2-carrying peptides generally have higher hydrophobicity but lower hydrophobic moment than those of + 3-carrying peptides. Therefore, the H and µ can be regarded as a compromise with respect to peptide charges and their distribution. In addition, the Trp seems to associate with high hydrophobicity as compared to Leu, while the Arg appears to have large hydrophobic moment relative to Lys (Zhou et al. 2013a, b). However, all the 56 designed heptapeptides were predicted to possess only a low (or moderately low) helicity by using the AGADIR algorithm (Muñoz and Serrano 1994), with h ranging between 7.6 and 51.7%. The Leu and Lys confer stronger helicity to peptides than Trp and Arg, respectively. This is basically in line with the amino acid intrinsic helical propensity observed in protein structures, which well meets the notion of rational peptide design (Tian et al. 2011, 2013, 2014).

Front view of the helical wheel of a standard α-helical heptapeptide, from which the residue distribution around the wheel is readily visualized, that is, in turn, X1 → X5 → X2 → X6 → X3 → X7 → X4
Hydrocarbon Stapling of Single-Turn Amphipathic α-Helical Heptapeptides
The 56 designed heptapeptides were found to possess a satisfactory profile of hydrophobicity and hydrophobic moment, but cannot effectively fold into standard α-helix in a spontaneous manner. Therefore, we considered employing hydrocarbon stapling technique to constrain the peptide helical conformation in water. The stapling was placed across the 2nd and 6th residues of heptapeptides, thus satisfying the (i, i + 4) rule for the span of an all-hydrocarbon bridge. In this respect, it is required that the 2nd and 6th residues cannot be positively charged amino acids; thus replacement of the two residues by nonpolar α-methyl,α-alkenylglycine would not change the physicochemical property of heptapeptides. In this consideration, we selected five from the 56 designed heptapeptides; they are structurally diverse and commercially available as well as possess relatively high hydrophobicity and hydrophobic moment. The five (unstapled) heptapeptides and their stapled counterparts are tabulated in Table 2, including three + 2-carrying peptides and two + 3-carrying peptides. Their MIC values were tested against antibiotic-sensitive S. aureus (ATCC25923) and methicillin-resistant S. aureus (MRSA) using susceptibility assays. As can be seen in Table 2, there is no essential difference of measured MIC values between ATCC25923 and MRSA for the same peptides, suggesting that the antibacterial mechanism of the designed AMPs is different to those of small-molecule antibiotics. As might be expected, stapling can moderately or considerably improve these peptide potencies by two–tenfold, with MIC change from, at most, > 100 μg/ml to < 10 μg/ml upon the stapling. In particular, the stapled p(3,7) peptide exhibited high potency against the two strains and substantial activity increase as compared to its unstapled counterpart (MIC = 8.3 and 14 μg/ml for stapled peptide versus 76 and 62 for unstapled peptide against ATCC25923 and MRSA, respectively) (Fig. 2).

CD spectra of stapled and unstapled p(3,7) peptides in 0% and 50% TFE solutions
The membrane permeabilization activities of five stapled peptide and their unstapled counterparts were determined using NPN uptake assays with E. coli strain UB1005. NPN, a neutral hydrophobic fluorescent probe, is normally excluded by the outer membrane but exhibits increased fluorescence intensity when it partitions into the outer membrane disrupted by antibacterial agents (Lv et al. 2014). The plots of NPN uptake by E. coli against varying peptide concentrations are shown in Fig. 3. It is revealed that, as might be expected (i) the stapled peptides exhibit generally higher membrane permeabilization activity than their unstapled counterparts, and (ii) the activity is basically consistent with peptide antibacterial potency, that is, a peptide with stronger antibacterial potency generally has higher permeabilization activity. For example, the stapled p(3,7) peptide was found to have the highest antibacterial activity with MIC value of 8.3 ± 1.8 μg/ml in all designed candidates; the peptide can also efficiently disrupt bacterial outer membrane with NPN uptake potency of 85.8% at concentration 10 μM. In contrast, its unstapled counterpart has only a moderate NPN uptake potency of 45.5% at the concentration.

NPN uptake by E. coli in the presence of varying peptide concentrations at 0, 1, 2, 5 and 10 μM. a stapled and unstapled p(1,5) peptides, b stapled and unstapled p(3,7) peptides, c stapled and unstapled p(7,4) peptides, d stapled and unstapled p(4,1,5) peptides, and e stapled and unstapled p(7,4,1) peptides
CD spectroscopy was used to characterize the secondary structure of five unstapled heptapeptides and their stapled counterparts in phosphate buffer, indicating a significant increase in peptide helical content upon the stapling, with helicity change from h = 14.1%–33.7% (for unstapled peptides) to h = 58.9–75.1% (for stapled peptides). In addition, we also compared the CD spectra of stapled and unstapled p(3,7) peptides in 0% and 50% TFE solutions (TFE:H2O = 0:100 and 50:50, respectively). As can be seen in Fig. 2, the unstapled peptide shows a typical spectral feature of random coil structure in 0% TFE solution (helicity = 26.3%), characterized by a single negative band below 210 nm. Addition of 50% TFE results in an increased conformational transition to α-helix (helicity = 51.4%), which presents two negative bands at 208 and 222 nm along with one positive band at 192 nm. In contrast, the stapled peptide behaves as a well-folded helical conformation in 0% TFE solution (helicity = 65.9%), and presence of 50% TFE has only a modest effect on the peptide helical content (helicity = 67.4%).
The stapled p(3,7) peptide was determined to have the strongest antibacterial potency and the highest permeabilization activity in all designed peptide candidates. Here, the peptide and its unstapled counterpart were initially assigned with helical and linear conformations, respectively, and placed in water ~ 40 Å away from the membrane center, which gradually approached to and were then immersed into membrane over MD simulations. The hydrocarbon stapling can influence the interaction dynamics substantially, particularly in the phase of peptide penetrating into membrane. The whole simulation includes four key time points (Fig. 4): point 0: simulation is start (0 ns); point 1: peptide touches on hydrophilic membrane surface; point 2: peptide penetrates into hydrophilic membrane surface; point 3: peptide transfers from hydrophilic membrane surface to hydrophobic membrane interior; point 4: simulation is end (500 ns). By visually examining the peptide conformations at each point it is revealed that the stapled peptide with restriction imposed by hydrocarbon bridge can well maintain in helical conformation over the whole simulation, whereas a significant conformational change was observed for its unstapled counterpart during the simulations. By comparing the dynamics trajectories of stapled and unstapled peptides, it is noticed that the stapling would not essentially influence phase [point 0 → 1] (in water) and phase [point 3 → 4] (in hydrophobic membrane interior), since the stapling does not alter the peptide electrostaticity and hydrophobicity that dominate the long-range membrane–peptide attraction in phase [point 0 → 1] and the short-range membrane–peptide interaction in phase [point 3 → 4], respectively. However, the stapling seems to considerably reduce the simulation time of phase [point 1 → 2] and phase [point 2 → 3]. In addition, it is revealed that, although the unstapled peptide is highly disorder in water, it appears to be partially folded as a helical conformation upon penetrating into the membrane and immersed in the membrane interior. As shown in Fig. 4, the peptide is fully linear at point 0, which would become slightly helical at points 1 and 2, and then further increase its helical content at point 3 to reach final helical state at point 4, imparting that the lipid environment can help the structured folding of unstapled peptide. This is consistent with the CD analyses that the unstapled peptide is largely disordered in water, but can be partially structured in amphipathic membrane, whereas the stapled peptide always keeps in structured helical state in both water and membrane environments.

Dynamics simulations of the membrane-interacting course of stapled p(3,7) peptide and its unstapled counterpart with lipid bilayer. The stapled and unstapled peptides are initially assigned with helical and linear conformations, respectively, and placed in water ~ 40 Å away from the membrane center, which gradually approach to and are then immersed into membrane during the simulations. The whole simulation includes four key time points: point 0: simulation is start (0 ns), point 1: peptide touches on hydrophilic membrane surface, point 2: peptide penetrates into hydrophilic membrane surface; point 3: peptide transfers from hydrophilic membrane surface to hydrophobic membrane interior; point 4: simulation is end (500 ns). The conformation and orientation of simulated peptide at the four points are also shown. The x-axis is MD simulation time and the y-axis represents the distance r between the center of mass of peptide and the center of lipid bilayer model
References
Abbassi F, Lequin O, Piesse C, Goasdoué N, Foulon T, Nicolas P, Ladram A (2010) Temporin-SHf, a new type of phe-rich and hydrophobic ultrashort antimicrobial peptide. J Biol Chem 285:16880–16892
Bai X, Chen X (2019) Rational design, conformational analysis and membrane-penetrating dynamics study of Bac2A-derived antimicrobial peptides against gram-positive clinical strains isolated from pyemia. J Theor Biol 473:44–51
Bai Z, Hou S, Zhang S, Li Z, Zhou P (2017) Targeting self-binding peptides as a novel strategy to regulate protein activity and function: a case study on the proto-oncogene tyrosine protein kinase c-Src. J Chem Inf Model 57:835–845
Barlow DJ, Thornton JM (1988) Helix geometry in proteins. J Mol Biol 201:601–619
Bird GH, Crannell WC, Walensky LD (2011) Chemical synthesis of hydrocarbon-stapled peptides for protein interaction research and therapeutic targeting. Curr Protoc Chem Biol 3:99–117
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: and N.log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
De Paola I, Pirone L, Palmieri M, Balasco N, Esposito L, Russo L, Mazzà D, Di Marcotullio L, Di Gaetano S, Malgieri G, Vitagliano L, Pedone E, Zaccaro L (2015) Cullin3-BTB interface: a novel target for stapled peptides. PLoS ONE 10:e0121149
Dinh TTT, Kim DH, Lee BJ, Kim YW (2014) De novo design and their antimicrobial activity of stapled amphipathic helices of heptapeptides. Bull Korean Chem Soc 35:3632–3636
Dinh TTT, Kim DH, Nguyen TQ, Lee BJ, Kim YW (2015) N-capping effects of stapled heptapeptides on antimicrobial and hemolytic activities. Bull Korean Chem Soc 36:2511–2515
Frecer V, Ho B, Ding JL (2004) De novo design of potent antimicrobial peptides. Antimicrob Agents Chemother 48:3349–3357
Fu J, Yang H, Wang J (2018) Computational design of the helical hairpin structure of membrane-active antibacterial peptides based on RSV glycoprotein epitope scaffold. Comput Biol Chem 73:200–205
Gautier R, Douguet D, Antonny B, Drin G (2008) HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics 24:2101–2102
Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1:2876–2890
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a unstapled constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Jochim AL, Arora PS (2009) Assessment of helical interfaces in protein-protein interactions. Mol Biosyst 5:924–926
Jochim AL, Arora PS (2010) Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem Biol 5:919–923
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom froce field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–21123
Kukol A (2009) Lipid models for united-atom molecular dynamics simulations of proteins. J Chem Theory Comput 5:615–626
Lee DL, Powers JP, Pflegerl K, Vasil ML, Hancock RE, Hodges RS (2004) Effects of single D-amino acid substitutions on disruption of beta-sheet structure and hydrophobicity in cyclic 14-residue antimicrobial peptide analogs related to gramicidin S. J Pept Res 63:69–84
Li Z, Yan F, Miao Q, Meng Y, Wen L, Jiang Q, Zhou P (2019a) Self-binding peptides: binding-upon-folding versus folding-upon-binding. J Theor Biol 469:25–34
Li Z, Miao Q, Yan F, Meng Y, Zhou P (2019b) Machine learning in quantitative protein-peptide affinity prediction: implications for therapeutic peptide design. Curr Drug Metab 20:170–176
Luo H, Du T, Zhou P, Yang L, Mei H, Ng H, Zhang W, Shu M, Tong W, Shi L, Mendrick DL, Hong H (2015) Molecular docking to identify associations between drugs and class I human leukocyte antigens for predicting idiosyncratic drug reactions. Comb Chem High Throughput Screen 18:296–304
Luong HX, Ngoan DK, Lee TMB (2017) Mono-substitution effects on antimicrobial activity of stapled heptapeptides. Arch Pharm Res 40:713–719
Lv Y, Wang J, Gao H, Wang Z, Dong N, Ma Q, Shan A (2014) Antimicrobial properties and membrane-active mechanism of a potential α-helical antimicrobial derived from cathelicidin PMAP-36. PLoS ONE 9:e86364
Migoń D, Neubauer D, Kamysz W (2018) Hydrocarbon stapled antimicrobial peptides. Protein J 37:2–12
Mishra B, Wang G (2012) The importance of amino acid composition in natural AMPs: an evolutional, structural, and functional perspective. Front Immunol 3:221
Mishra B, Lushnikova T, Golla RM, Wang X, Wang G (2017) Design and surface immobilization of short anti-biofilm peptides. Acta Biomater 49:316–328
Muñoz V, Serrano L (1994) Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol 1:399–409
Ren Y, Chen X, Feng M, Wang Q, Zhou P (2011) Gaussian process: a promising approach for the modeling and prediction of peptide binding affinity to MHC proteins. Protein Pept Lett 18:670–678
Scholtz JM, Baldwin RL (1992) The mechanism of α-helix formation by peptides. Annu Rev Biophys Biomol Struct 21:95–118
Shepherd NE, Hoang HN, Abbenante G, Fairlie DP (2005) Single turn peptide alpha helices with exceptional stability in water. J Am Chem Soc 127:2974–2983
Tian F, Lv Y, Zhou P, Yang L (2011) Characterization of PDZ domain-peptide interactions using an integrated protocol of QM/MM, PB/SA, and CFEA analyses. J Comput Aided Mol Des 25:947–958
Tian F, Tan R, Guo T, Zhou P, Yang L (2013) Fast and reliable prediction of domain-peptide binding affinity using coarse-grained structure models. Biosystems 113:40–49
Tian F, Yang C, Wang C, Guo T, Zhou P (2014) Mutatomics analysis of the systematic thermostability profile of Bacillus subtilis lipase A. J Mol Model 20:2257
Tossi A, Sandri L, Giangaspero A (2000) Amphipathic, α-helical antimicrobial peptides. Biopolymers 55:4–30
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718
Walensky LD, Bird GH (2014) Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem 57:6275–6288
Wang Y, Zhao T, Wei D, Strandberg E, Ulrich AS, Ulmschneider JP (2014) How reliable are molecular dynamics simulations of membrane active antimicrobial peptides? Biochim Biophys Acta 1838:2280–2288
Wang G, Li X, Wang Z (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44:D1087–D1093
Wang J, Zhang J, Sun X, Liu C, Li X, Chen L (2019) Molecular design of sequence-minimized, structure-optimized, and hydrocarbon-stapled helix-helix interactions in the trimer-of-hairpins motif of pediatric pneumonia RSV-F protein. Chem Biol Drug Des 94:1292–1299
Yang C, Zhang S, He P, Wang C, Huang J, Zhou P (2015a) Self-binding peptides: folding or binding. J Chem Inf Model 55:329–342
Yang C, Wang C, Zhang S, Huang J, Zhou P (2015b) Structural and energetic insights into the intermolecular interaction among human leukocyte antigens, clinical hypersensitive drugs and antigenic peptides. Mol Simul 41:741–751
Yang C, Zhang S, Bai Z, Hou S, Wu D, Huang J, Zhou P (2016) A two-step binding mechanism for the self-binding peptide recognition of target domains. Mol. Biosyst 12:1201–1213
Yang R, Zhang G, Zhang F, Li Z, Huang C (2018) Membrane permeabilization design of antimicrobial peptides based on chikungunya virus fusion domain scaffold and its antibacterial activity against Gram-positive Streptococcus pneumoniae in respiratory infection. Biochimie 146:139–147
Yau YH, Ho B, Tan NS, Ng ML, Ding JL (2001) High therapeutic index of factor C Sushi peptides: potent antimicrobials against Pseudomonas aeruginosa. Antimicrob Agents Chemother 45:2820–2825
Ye H (2018) Molecular design of antimicrobial peptides based on hemagglutinin fusion domain to combat antibiotic resistance in bacterial infection. J Pept Sci 24:e3068
Yu H, Zhou P, Deng M, Shang Z (2014) Indirect readout in protein-peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model 54:2022–2032
Zhou P, Wang C, Tian F, Ren Y, Yang C, Huang J (2013a) Biomacromolecular quantitative structure-activity relationship (BioQSAR): a proof-of-concept study on the modeling, prediction and interpretation of protein-protein binding affinity. J Comput Aided Mol Des 27:67–78
Zhou P, Yang C, Ren Y, Wang C, Tian F (2013b) What are the ideal properties for functional food peptides with antihypertensive effect? A computational peptidology approach. Food Chem 141:2967–2973
Zhou P, Zhang S, Wang Y, Yang C, Huang J (2016) Structural modeling of HLA-B1502 peptide carbamazepine T-cell receptor complex architecture: implication for the molecular mechanism of carbamazepine-induced Stevens-Johnson syndrome toxic epidermal necrolysis. J Biomol Struct Dyn 34:1806–1817
Zhou P, Hou S, Bai Z, Li Z, Wang H, Chen Z, Meng Y (2018) Disrupting the intramolecular interaction between proto-oncogene c-Src SH3 domain and its self-binding peptide PPII with rationally designed peptide ligands. Artif Cells Nanomed Biotechnol 46:1122–1131
Zhou P, Miao Q, Yan F, Li Z, Jiang Q, Wen L, Meng Y (2019) Is protein context responsible for peptide-mediated interactions? Mol Omics 15:280–295
Acknowledgments
This work was funded by the YCH foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, Z., Yu, X., Zhang, A. et al. De Novo Hydrocarbon-Stapling Design of Single-Turn α-Helical Antimicrobial Peptides. Int J Pept Res Ther 26, 1711–1719 (2020). https://doi.org/10.1007/s10989-019-09964-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-019-09964-7