
Abstract
[Ca2+] transients inside the sarcoplasmic reticulum (SR) were recorded in frog skeletal muscle twitch fibers under voltage clamp using the low affinity indicator Mag Fluo 4 (loaded in its AM form) with the purpose of studying the effect on Ca2+ release of extrinsic Ca2+ buffers (i.e. BAPTA) added at high concentration to the myoplasm. When the extrinsic Ca2+ buffer is added to the myoplasm, part of the released Ca2+ binds to it, reducing the Ca2+ signal reported by a myoplasmic indicator. This, in turn, hinders the quantification of the amount of Ca2+ released. Monitoring release by measuring [Ca2+] inside the SR avoids this problem. The application of extrinsic buffers at high concentration reduced the resting [Ca2+] in the SR ([Ca2+]SR) continuously from a starting value close to 400 μM reaching the range of 100 μM in about half an hour. The effect of reducing resting [Ca2+]SR on the Ca2+ permeability of the SR activated by voltage clamp depolarization to 0 mV was studied in cells where the myoplasmic [Ca2+] ([Ca2+]myo) transients were simultaneously recorded with Rhod2. The Ca2+ release flux was calculated from [Ca2+]myo and divided by [Ca2+]SR to obtain the permeability. Peak permeability was significantly reduced, from 0.026 ± 0.005 ms−1 at resting [Ca2+]SR = 372 ± 5 μM to 0.021 ± 0.004 ms−1 at resting [Ca2+]SR = 120 ± 16 μM (n = 4, p = 0.03). The time averaged permeability was not significantly changed (0.009 ± 0.003 and 0.010 ± 0.003 ms−1, at the higher and lower [Ca2+]SR respectively). Once the cells were equilibrated with the high buffer intracellular solution, the change in [Ca2+]SR (Δ[Ca2+]SR) in response to voltage clamp depolarization (0 mV, 200 ms) in 20 mM BAPTA was significantly lower (Δ[Ca2+]SR = 30.2 ± 3.5 μM from resting [Ca2+]SR = 88.8 ± 13.6 μM, n = 5) than in 40 mM EGTA (Δ[Ca2+]SR = 72.2 ± 10.4 μM from resting [Ca2+]SR = 98.2 ± 15.6 μM, n = 4) suggesting that a Ca2+ activated component of release was suppressed by BAPTA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In skeletal muscle, the Ca2+ that activates contraction is released from the terminal cisternae (TC) of the sarcoplasmic reticulum (SR). The pathways for release are the ryanodine receptor channels (RyRs) present in the TC (Sutko and Airey 1996; Fill and Copello 2002). In mammals the predominant isoform of RyRs is termed type 1. Another type, RyR3, is present only in some muscles and even there their expression is less than that of RyR1. The RyR1s are arranged in a double row at the junctional face of the TC, where it comes in apposition with the membrane of the transverse tubule (TT) (Block et al. 1988). In the TT membrane there is a set of molecules that interact with RyRs, the dihydropyridine receptors (DHPRs), L-type Ca2+ channels that serve as voltage sensors for excitation–contraction coupling (ECC) (Ríos and Pizarro 1991). The DHPRs are also organized in a double row of tetrads, the center to center distance between tetrads is twice that between the RyRs. The double row in the junctional TC is orthogonal, the elements in one row are exactly aligned with those in the other row. The double row in TT is in zigzag, the elements in one row are shifted half a period respect to the elements in the other row. Thus, only one every other RyR faces a tetrad of DHPRs.
Based on the structure described by Block et al. (1988), it was proposed that those RyRs that do not face a DHPR tetrad might be activated by Ca2+ released from the SR through the RyRs activated by the DHPRs (Ríos and Pizarro 1988). This mechanism, named Ca2+ induced Ca2+ release (CICR), was originally described in mechanically skinned fibers (Endo et al. 1970) and its role in the physiological release process has been unclear ever since. The crucial experiment to prove a physiological CICR role is to add a rapid Ca2+ buffer to prevent Ca2+ binding to myoplasmic sites responsible for the activation of the SR Ca2+ release channels by CICR. This condition is attained by adding the fast chelator BAPTA at high concentration to the intracellular milieu. This type of experiment has been performed by different groups, yielding contradictory evidence for CICR. Jacquemond et al. (1991) and Csernoch et al. (1993) reported that the Ca2+ permeability of the SR was reduced by BAPTA injection (3.8 mM). BAPTA suppressed the early peak of the permeability but spared the steady level attained at 100 ms. These authors concluded that the effect of BAPTA was due to inhibition of CICR, which was responsible for the peak.
On the other hand, Jong et al. (1993) and Jong et al. (1995) concluded that the permeability was enhanced as consequence of the increased Ca2+ buffering in the presence of 2–3 mM Fura2 (a BAPTA derivative) in the myoplasm. The peak was eliminated, the permeability increased during the pulse up to a steady level that was higher than the peak observed previous to the application of Fura2. Their conclusion was that increased Ca2+ buffering suppressed Ca2+ dependent inactivation and potentiated the permeability of the SR.
Both groups agreed that the time course of the SR Ca2+ permeability in the presence of a fast Ca2+ buffer consisted in a monotonic rise towards a steady level. They differed in the estimation of its size.
There are not obvious reasons for the discrepancy but many methodological factors might be responsible. It might be the result of measuring the myoplasmic [Ca2+] transients in low and high buffer with different indicators with very different dissociation constants (kd), as in the case of Jong et al. (1993). The [BAPTA] attained after injection could have been underestimated, as in the case of Jacquemond et al. (1991) and Csernoch et al. (1993). Both studies corrected for change in driving force during release under the hypothesis that the flux was proportional to the total [Ca]SR. A fact that might have also contributed to the different outcomes was the different method to obtain the resting total [Ca]SR in the two studies.
Pape et al. (2002) and Fenelon and Pape (2002) revisited the effect of BAPTA on the SR Ca2+ permeability in frog cut fibers under voltage clamp with a different method. In the presence of 20 mM EGTA, to presumably capture all the released Ca2+, the change in [H+] in the myoplasm that resulted from the displacement of H+ from EGTA upon Ca2+ binding was measured with phenol red. The proton signal measured the amount of Ca2+ bound to EGTA. The addition of 8 mM BAPTA to the myoplasm captured a fraction of the released Ca2+. As this chelator did not release as many protons upon binding Ca2+ (only 10% compared to EGTA) the phenol red signal was trivially reduced. Under the assumption that the free Ca2+ was below the kds of both chelators, the authors argued that the CaEGTA signal measured in BAPTA was still linearly related to the total Ca2+ released. Using a rather indirect method, they concluded that BAPTA suppressed the SR Ca2+ permeability in a voltage dependent manner. While at − 60 mV, the permeability was not diminished by BAPTA, a 50% inhibition was found at − 45 mV and presumably at more depolarized voltages. Therefore, they concluded that a CICR component was activated at depolarized voltage, positive to − 60 mV. These latter studies broadly agreed with those of Jacquemond et al. (1991) and Csernoch et al. (1993).
In all the studies described above, the Ca2+ signal reported by the myoplasmic indicator is reduced because part of the Ca2+ binds to the added buffer. In order to estimate the total Ca2+ released, the Ca2+ bound to buffer has to be estimated. In all these studies, the result depends heavily on this estimation.
An isoform equivalent to RyR1 named α is present in the amphibian, avian and fish skeletal muscles. A second isoform named β is present (Sutko and Airey 1996; Murayama and Kurebayashi 2011). These RyRs β, equivalent to RyRs 3, are extra junctional. They are organized in shorter double rows in zigzag on the lateral, non junctional, faces of the TC. These channels do not interact with the DHPR (Felder and Franzini-Armstrong 2002). Their physiological activation—presumed to be by myoplasmic Ca2+—generates the Ca2+ sparks seen in frog but not in mammalian muscle cells under normal conditions (Ríos 2018). Sparks are also generated by the cardiac isoform of RyR and are activated by CICR, the ECC mechanism of the heart (Cheng and Lederer 2008).
Despite the important contribution of Ca2+ sparks to the release process, a role of CICR in the physiological ECC of amphibian skeletal muscle is not generally accepted (Endo 2009; Murayama and Kurebayashi 2011). A different opinion is held by Ríos (2018), who concluded that the overall evidence supported the involvement of CICR in the amphibian.
All the studies described above were carried out by monitoring the myoplasmic Ca2+ transient elicited by the depolarization of the cell membrane voltage. More recently, recording of the [Ca2+] in the SR ([Ca2+]SR) has become possible. The intra SR Ca2+ transients were studied either with membrane permeable dyes that reach the SR lumen through the SR membrane (Kabbara and Allen 2001; Launikonis et al. 2006; Pape et al. 2007; Ziman et al. 2010; Robin et al. 2012) or with genetically coded probes targeted to the SR (Rudolf et al. 2006; Canato et al. 2010; Sztretye et al. 2011a; Tang et al. 2011). In these studies, resting [Ca2+]SR ranging from 300 to 1000 μM were reported.
[Ca2+]SR transients in frog twitch fibers under voltage clamp in a double Vaseline gap were studied by Pape et al. (2007) and Fénelon et al. (2012). Based on combined measurements in the SR (with tetramethilmurexyde (TMX), a membrane permeant indicator) and the myoplasm (with the phenol red- EGTA method) they obtained the time course of the Ca2+ permeability of the SR activated by voltage clamp pulses (Fenelon et al. 2012). Their conclusion was that there was no luminal regulation of the release channels and that the permeability reduction observed at very low [Ca2+]SR was due to reduction of CICR, a mechanism that acts on the myoplasmic side of the release channels.
This is in sharp contrast with the interpretation of Sztretye et al. (2011b) of a study in mouse skeletal muscle. These authors measured SR and myoplasmic Ca2+ signals in isolated myocytes under whole cell patch clamp. For the intra SR measurements, a biosensor based on CSQ was used while the myoplasmic transient was monitored with X-Rhod1. By comparing wild type and CSQ knock out mice, they concluded that the voltage activated permeability was reduced by CSQ as a consequence of the reduction in [Ca2+]SR.
A complex process of modulation by CSQ on the luminal side of the RyR1 has been described in reconstituted mammalian channels (Beard et al. 2004). The absence or less prominent modulation by CSQ in amphibians might reflect species differences. For instance, Donoso et al. (1995) found that the dependence on luminal [Ca2+] of the rate constant of release was weaker in frog triads than in their rabbit counterparts.
The intra SR recording provides a way to study the effect of extrinsic Ca2+ buffers (i.e. BAPTA) added in high concentration to the myoplasm without the complication derived of monitoring release with a myoplasmic indicator, as it was previously discussed.
With this aim, we implemented the measurement of the intra [Ca2+]SR transients with the low affinity indicator Mag Fluo 4. In some experiments, we simultaneously measured the myoplasmic [Ca2+] ([Ca2+]myo) transients with Rhod2 or RhodFF.
When extrinsic buffers at high concentration were applied (20 or 40 mM EGTA, 10 or 20 mM BAPTA) the resting [Ca2+]SR continuously decreased.
We studied the permeability of the SR activated by voltage clamp depolarization to 0 mV at different resting [Ca2+]SR. It was found to be rather independent of the [Ca2+]SR in the range between 400 and 100 μM, although important kinetic differences were observed..
The main observation of this study was that - once the cells were equilibrated with the high buffer intracellular solution—the change in [Ca2+]SR (Δ[Ca2+]SR) in response to voltage clamp depolarization in BAPTA was lower than in EGTA. This result supports Ca2+ activated component suppressed by BAPTA.
Methods
The experiments were carried out on frog (Rana catesbeiana) cut fibres voltage clamped in a two-Vaseline gap apparatus (Kovacs et al. 1983). After sedation in 5% ethanol and double pithing, frogs were decapitated following a procedure approved by the Honorary Committee for Animal Experimentation (CHEA) of our institution. 2 cm-long pieces of fiber were dissected from the semitendinosus muscle and mounted in a three compartment Lucite chamber where the double Vaseline gap was made. The fibers were mounted at slack length and contraction was prevented by the extracellular application of 100 μM n-Benzyl-p-Toluenesulfonamide (BTS). Several notches were made to the regions of the fiber in both end compartments in order to allow exchange of intracellular solution. The preparation was cooled to temperatures between 12 and 15 °C by means of a Peltier device. Membrane currents and optical signals were recorded and analyzed as previously described (De Armas et al. 1998) and briefly summarized below.
Solutions
The extracellular solution consisted of 130 mM Tetraethylammonium methansulfonate (TEACH3SO3), 8 mM CoSO4 and 10 mM Tris maleate. To further suppress ionic currents, ion channel blockers were added as follows: 0.05 μM TTX, 1 mM Anthracene- 9- Carboxilic (A9C) acid and 1 mM 3,4-Diaminopyridine. All external solutions were titrated to pH 7.0 with TEAOH.
The intracellular solution consisted of 120 mM Cs-glutamate, 10 mM Cs Tris maleate and 5 mM MgATP. Ethylene glycol-bis(b-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) was added to the internal solution at 1, 5, 20 or 40 mM adding the necessary amount of CaCl2 in order to yield a free [Ca2+] = 20 nM, with an apparent dissociation constant (kd) of 0.37 µM for EGTA, calculated with the program Maxchelator for pH 7.0, temperature of 15 °C and 0.15 M ionic strength. In some experiments 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) instead of EGTA was applied at a concentration of 10 or 20 mM. Free Ca2+ was set at the same level, calculated using Maxchelator as described. Rhod2 or RhodFF (tripotassium salt, Molecular Probes, Eugene, OR, USA) were used as Ca2+ indicators, added to the intracellular solution at 0.4 mM. The pH of the intracellular solution was titrated to 7.0 with Cs OH.
Cell loading with Mag Fluo4AM or Fluo5N AM
For loading into the cells, the indicators were dissolved in Ringer solution at a final concentration of 50 μM. The indicators were previously dissolved in a 20% solution of Pluronic acid in DMSO and added to the Ringer solution. The final dilution of DMSO in the loading solution was 5 in 1000. To load the AM indicator, the muscle was dissected into two or three bundles after removal of the connective tissue sheaths. When Mag Fluo4 AM (Molecular Probes, Eugene, OR, USA) was used, these bundles were kept in the loading Ringer solution for one hour at room temperature. After that, the muscle was rinsed and placed in normal Ringer for at least two more hours at room temperature before dissection and mounting of single fibers. This period was important, not only for complete deesterification but for the removal of the myoplasmic dye. If shorter periods after loading were used (30 min), the fibers showed a clear early positive going myoplasmic signal followed by the negative going SR component. Fluo 5N AM was loaded for 30 min at 30 °C, after that the muscle was kept for half an hour at room temperature in normal Ringer before dissecting and mounting the single fibers in the experimental chamber. The loading with Fluo 5N was always weaker than with Mag Fluo 4. Repeated attempts to load at room temperature, as described for Mag Fluo4 AM, were not successful.
Electronics and data acquisition
Data acquisition was performed with a 16-bit resolution, 100 kHz board (HSDAS-16, Analogic Corporation, Wakefield, MA, USA). Sampling rates were 20 kHz per channel. Data was compressed by averaging and stored at 0.2, 1 or 2 ms per point depending on the length of the recording. Signals were filtered by a one-pole passive filter with 0.5 kHz cut-off frequency prior to acquisition by the A-D converter. Command pulses were generated with the D/A channels of the HSDAS board. Membrane currents, membrane voltage, and emitted light were measured simultaneously during fiber activation by clamp pulses.
Optical measurements and processing of the optical signals
To measure myoplasmic Ca2+ signals, Rhod-2 or Rhod FF were used. Rhod-2 was used in the experiments with internal solution containing 5 mM EGTA or higher. The myoplasmic [Ca2+] transient was derived from the fluorescent signal of Rhod2, as described by Shirokova et al. (1996) for Fluo3.
The concentration of Rhod2 was obtained by measuring the absorbance at 540 nm using an extinction coefficient of ε560 = 5.92 × 104 M−1 cm−1 obtained in our apparatus. The absorbance readings were taken periodically, every 10–20 min, and linearly interpolated to estimate the concentration of the indicator at the time of measurement of each transient. The rate constants of Rhod 2 were koff = 110 s−1 and kon = 90 s−1 µM−1 as described elsewhere (Olivera and Pizarro 2016), resulting in a kd = 1.22 μM.
In a series of experiments, cells were studied with a 1 mM EGTA internal solution. In this case we used the low affinity indicator Rhod FF (kd = 19 μM, given by the manufacturer, Teflabs, CA, USA) because Rhod2 tended to saturate when the myoplasmic Ca2+ buffering was low, especially during long lasting (several seconds) pulses to 0 mV. Rhod FF was assumed in equilibrium with Ca2+. The fluorescence calibration parameters were measured in vitro in our optical apparatus.
To measure the signals within the SR, either Mag-Fluo4 or Fluo 5N were used. Mag Fluo4 worked better in our hands, and therefore was routinely used. A series of experiments with Fluo 5N were carried out to compare the signals from the two indicators to detect Mg2+ dependent contribution to the signal reported by Mag Fluo4. We did not find systematic differences between the two indicators. The calibration procedures for the indicators within the SR will be described in the “Results” section.
The dyes were excited by epi-illumination with LED based light sources, centered at 490 and 535 nm (Cool Led, UK) arranged at 90°. The optical apparatus implemented in a right-side up epifluorescence microscope (Leitz Laborlux) is schematically represented in Fig. 1. The light from the 535 nm source went through a dichroic mirror (DM1) that reflected the light coming from the 490 nm source towards the epi-illumination system. Both sources were turned on simultaneously right before the acquisition started and were turned off immediately after it was completed. The illuminating light passed through a double band interference filter (IFX, with center wavelengths ± half bandwiths of 480 ± 20 and 550 ± 10 nm) placed between the light sources and a double band dichroic mirror (DM2) where the excitation light was reflected. The emitted light went through DM2 (530 ± 15 and 600 ± 25 nm) and was filtered at 530 ± 15 and 590 ± 20 nm by a second double band interference filter (IFM). The light emerging from IFM was split by a dichroic mirror (DM3) that reflected light below 560 nm and allowed the higher wavelengths to go through. All the optical components were purchased from Chroma technology, USA. The signals were recorded by two high sensitivity photodiodes PD1 and 2 (HUV-2000, EG&G, Quebec, Canada).

The schematics of the optical apparatus implemented in a right-side up epi fluorescence microscope. The indicators (Rhod-2 and Mag-Fluo4 or Fluo 5N) were excited by epi-illumination by two LED based light sources centered at 490 and 535 nm arranged at 90°. The light from the 535 nm source passed through a dichroic mirror (DM1) that reflected the light coming from the 490 nm source towards the epi-illumination system. The illuminating light passed through a double band interference filter (IFX, 480 and 550 nm) placed between the light sources and a double band dichroic mirror (DM2). The emitted light went through the dichroic mirror and was filtered at 530 and 590 nm by a second double band interference filter (IFM) and splitted by a dichroic mirror (DM3) that reflected light below 560 nm and allowed the higher wavelengths to go through. The signals were recorded by two high sensitivity photodiodes PD1 and 2
Calculation of Ca2+ release flux
Ca2+ release flux was calculated from the [Ca2+]myo transients with the method of Melzer et al. (1987) as the sum of the rate of increase of free [Ca2+]myo and the rate of removal by an explicit removal model. The removal model included Ca2+ buffers known to be present in the myoplasm (parvalbumin, troponin) plus those added to the internal solution (i.e. EGTA and dye) as well as Ca2+ pumping to the SR. This model was fitted to the decay of a set of [Ca2+] myo transients elicited by pulses of various amplitudes and durations, assuming that during the fit the release flux was null. The fit was repeated at different times during the experiment. Some of the parameters characterizing the Ca2+ binding sites were adjusted in the fit and the values obtained are given in the figure legends. The remaining parameters were kept constant with values given by Brum et al. (1988a) or by González and Ríos (1993). The choice of parameters to be adjusted was rather arbitrary. Since EGTA was added to the internal solution at a rather high concentration, its concentration or one of its rate constants was always one of the adjustable parameters in the fits. The concentration of parvalbumin and the maximum pump flux were also routinely adjustable parameters. The values obtained in the fit do not necessarily reflect the actual properties of each Ca2+ binding site but the success of the removal model as a whole to reproduce the decay of the [Ca2+]myo transients after the end of voltage clamp pulses of various amplitudes and durations. The reliability of the method has been shown by Pizarro and Ríos (2004). The waveform obtained, loosely referred as Ca2+ release flux, is equal to the rate of change in total [Ca2+]myo (free plus bound) plus the rate at which Ca2+ is pumped back to the SR.
Measurement of intramembranous charge movement current
Asymmetric currents were obtained as the difference between the current during the test pulses and the current measured during control pulses, the latter was scaled by the ratio between the amplitude of the test pulse and the amplitude of the control pulse. Charge movement transients were obtained by subtraction of sloping baselines, separately fitted to the last 50 ms of ON and OFF portions of the record. Test pulses were applied from the holding potential of − 80 mV or more negative. The control pulses were 20 mV pulses, applied to − 100 mV from a potential of − 120 mV where the membrane was held for a short period after the acquisition of the test pulse. Usually, 4 to 8 control pulses were applied for every test pulse. The currents during the controls were averaged before subtraction. Charge displacements QON and QOFF were obtained by integration of the corresponding transients and normalized to the linear capacitance measured in the control pulses.
Statistics
All data is given as mean ± standard error of the mean (s. e. m.). Statistical significance was estimated by t-test (either paired or independent) at p ≤ 0.05.
Results
Simultaneous recording of myoplasmic and intra-SR Ca2+ transients
In experiments with only one dye at a time, it was found that Mag Fluo 4 fluorescence above 560 nm (both the resting fluorescence and the transient signal elicited upon fiber activation) was about 75% of the one measured below 560 nm. On the other hand Rhod2 fluorescence below 560 nm was small, only 1% the signal recorded above that wavelength. Thus the light below 560 nm, a signal that will be termed S1, was mostly due to Mag Fluo 4 fluorescence. When Rhod2 was present, the small signal S1 only became important if the concentration was high.
When both dyes were present in the cell, the signal S2 (the light above 560 nm) was a combination of the Rhod-2 and Mag Fluo-4 emission. As it was already mentioned, the contribution of Mag Fluo4 to the signal recorded in the upper detector was substantial, therefore to obtain a pure Rhod2 signal it has to be subtracted after proper scaling.
S1 and S2 are the raw signals that are directly acquired as outputs of the PD1 and PD2 photodetectors, respectively.
Calling MF the Mag Fluo 4 signal below 560 nm and R the Rhod2 signal above that wavelength, we have
Solving the two simultaneous equations for R and F yields
Figure 2 shows the acquired and the subtracted signals in response to steps of 0 mV. In Fig. 2a, c the raw signals recorded when the concentration of Rhod2 was low. In this condition, the signal S1 was basically all due to Mag Fluo 4 fluorescence. As shown in Fig. 2b, after subtraction, the signal MF was basically identical to S1. Figure 2d shows the pure Rhod2 signal (R) at the upper detector.

Optical records in the presence of the two dyes, Rhod 2 in the myoplasm and Mag Fluo 4 in the sarcoplasmic reticulum. In panels a and c the raw signals recorded with a low concentration of Rhod2 are shown. In a the signal S1 is the emitted light below 560 nm. This is basically all due to Mag Fluo 4, as shown in b after subtraction (signal MF, as explained in the text) the signal is identical. Panel c shows the raw signal (S2) recorded by the upper detector, consisting of wavelengths above 560 nm. d shows the pure Rhod2 signal (R), obtained after subtraction. Records obtained in the same cell 42 min later are shown in the four lower panels. By this time the Rhod 2 concentration had increased substantially (from 55 to 439 μM) and the Mag Fluo 4 fluorescence was reduced probably by reduction of the free [Ca2+]SR as a consequence of the application of 40 mM EGTA to the intracellular solution (35 min earlier). e shows record S1, obtained in the presence of high [Rhod2]. This record has a small early positive going signal superimposed to the Mag Fluo 4 signal which is corrected after subtraction, signal labeled MF shown in f. g shows the simultaneously recorded S2. In g the signal R, obtained after subtraction, is shown. Fiber 171,116. Diameter 112 μm, optical path 90 μm
The four lower panels show records obtained later in the same cell. By this time, the Rhod 2 concentration had substantially increased. Figure 2E shows S1 in the presence of high concentration of Rhod2, a small early positive going signal was superimposed to the Mag Fluo 4 signal. The Mag Fluo 4 resting fluorescence was reduced, probably due to a reduction of [Ca2+]SR. Figure 2F shows the MF signal obtained with Eq. 1d, the early component was corrected after subtraction.
The record S2 is shown in Fig. 2G and the calculated R signal is shown in Fig. 2H.
The subtraction procedure depended on the accuracy of the scaling factor for the Mag Fluo 4 signal. As we always started the experiment with the cell loaded with Mag Fluo 4 (before adding Rhod2) the ratio between the signals was measured in each cell. This compensated for the small error that might arise due to change in the aligning of the photo detectors between experiments.
Calibration of the SR signals
The signal measured when only the AM form of MagFluo4 (or Fluo5N) was used originated in the dye within the SR. Upon voltage clamp depolarization, the signal was always a reduction in fluorescence. This was as expected if the indicator was present inside a compartment from which Ca2+ was released. The negative going signals observed in the presence of low Ca 2+ buffering in the myoplasm ([EGTA] ≤ 1 mM) strongly supported the notion that the indicator concentration in the myoplasm was very low. The amplitude of the fluorescence signal, or the amplitude of the Δ[Ca2+]SR calculated from it, had a sigmoidal voltage dependence that correlated well with the voltage dependence of the simultaneously measured charge movement.
To calibrate the signal, we measured in vitro the fluorescence ratio between the Ca saturated and the Ca free form of the indicator (Fmax/Fmin). This was done in a 112 μm wide glass capillary placed in our optical apparatus and excited at the same intensity as in the experiments. The form of the dye used for the calibration (either 100 μM of Mag Fluo 4 K3 or Fluo 5 N K5) was dissolved in our internal solution. An average ratio of 14 was measured for Mag Fluo4 and used throughout. Fluo 5N gave us always a ratio above 100, but its determination was erratic due to the very faint fluorescence at zero Ca2+. Since we did not use this latter dye extensively, we did not pursue the issue any further.
The kdDye of MagFluo4 was determined by fitting a one to one binding equation to the fluorescence measurements at free [Ca2+] values 0 μM, 20 μM, 100 μM, 10 mM. We found a value of 90 μM that, although higher than the one given by the manufacturer, is similar to the one reported by Hollingworth et al. (2009). Finally, an estimate of the minimum fluorescence (Fmin) was needed. In principle, this parameter would depend on the concentration of dye achieved during the loading procedure as well as on geometrical factors. Therefore, it was necessary to determine Fmin in each cell. We did so, at the end of the experiment, by pharmacologically depleting the SR in the presence of high Ca2+ buffering in the cytoplasm.
After the application of a 40 mM EGTA Ca-free internal solution to the cut ends, followed by enough time for equilibration with the myoplasm, either 0.1 mM 4 Cl m-cresol or 10 mM caffeine plus 50 μM CPA was extracellularly applied to release Ca2+ from the SR. The released Ca2+ would bind to EGTA, minimizing any increase of fluorescence due to dye in the myoplasm or in organelles other than the SR. The remaining fluorescence would be mostly due to the dye in the SR, mainly to the fraction bound to Mg. This minimum fluorescence in the cell was assumed to come from the SR and was our estimate of Fmin. The complete depletion of the SR was tested by voltage clamp pulses. If the pulse did not elicit a signal, this was taken as indication of full depletion. Assuming that the Fmax/Fmin ratio and the kdDye were the same in the cell as in vitro, the maximum fluorescence was estimated and the free [Ca2+]SR was calculated with Eq. (2).
Figure 3 shows an example of the calibration of the MagFluo4 signal. Figure 3a shows the raw fluorescence of MagFluo4 measured in the same cell. Records 1 and 2 are transients in response to pulses to 0 mV. Record 1 was taken early during the experiment, when the internal solution contained 1 mM EGTA. Record 2 was acquired later, 47 min after the application of 40 mM EGTA to the internal solution. Records 3 and 4 are fully depleted and background fluorescence (measured with the fiber moved out of the illuminated field) respectively, the trace labeled 5 is the value of the maximum fluorescence calculated by multiplying the background subtracted fully depleted fluorescence by the Fmax/Fmin ratio. Figure 3b shows the [Ca2+]SR signals calculated from those in panel a.

The calibration of the Mag Fluo4 signal, only this dye was present in this cell. a shows the raw fluorescence of Mag Fluo4. Records 1 and 2 are transients in response to pulses to 0 mV, 1 taken early during the experiment in 1 mM EGTA internal solution, 2 was taken later on, after the application for 47 min of a 40 mM EGTA internal solution. Records 4 and 4 are fully depleted and background fluorescence respectively, 5 is the value of the maximum fluorescence calculated by multiplying background subtracted fully depleted fluorescence by the ratio. b are the [Ca] SR obtained with Eq. 3 as explained in the text, records 1 and 2 correspond to the records in 3A indicated by the same numbers. Fiber 280,515
Of the two dyes that were successfully loaded into the SR, only Mag Fluo 4 gave a reliable calibration because Fmin could be robustly distinguished from the background fluorescence. We think that this was due to the Mg2+ bound form of the indicator that was rather substantial at our estimated 0.6 mM free [Mg2+]. Fluo 5N minimum fluorescence was very weak and almost indistinguishable from the background. Therefore the signals were not calibrated.
Figure 4 shows records with MagFluo4 and Fluo5N from two different cells. In Fig, 4 A the fluorescent signals obtained with MagFluo4 are shown and in B with Fluo5N. Both were obtained first with an internal solution with 1 mM EGTA and after the application of 40 mM EGTA (traces labeled1 and 2, respectively). Note that in the latter case the resting fluorescence and presumably the free [Ca2+]SR was drastically reduced. The steady traces at the bottom were the minimum fluorescence and background. The signals with both dyes were very similar, suggesting that there was no contribution of Mg2+ to the transients recorded with MagFluo4.

Records with Mag Fluo 4 and Fluo 5 N from two different cells with only one dye at a time. In a, at the top is the voltage clamp pulse. Below it, the fluorescent signals obtained with Mag Fluo 4 are shown. In b an experiment with Fluo 5 N is depicted. At the top the voltage clamp pulse is plotted, below it, the fluorescent signals. In both cases the records labeled 1 were obtained with an internal solution with 1 mM EGTA and those labeled 2 after the application of 40 mM EGTA. Note that, in 40 mM EGTA, the resting fluorescence and presumably the free [Ca2+] was drastically reduced in both cases. The steady traces at the bottom were the minimum fluorescence and background measured with the fiber moved out of the illuminated field. The signals with both dyes were very similar suggesting that there was no contribution of Mg2+ to the transients recorded with Mag Fluo4. Fibers: 280,515 in panel A and 020515 in B
The MagFluo4 minimum fluorescence might be over estimated by the contribution to it by other sources, mainly fluorescence by dye trapped in mitochondria. Therefore the maximum fluorescence would be proportionally overestimated, increasing the value of the denominator in Eq. (2). Therefore the [Ca2+]SR could be underestimated. Our estimate of the kdDye could be also undervalued, as it is known that in the intracellular environment the kd measured for other similar dyes was increased compared to the one obtained in vitro. This would also contribute to an underestimation of the free [Ca2+]SR.
Time course of the SR Ca2+ signal during long lasting depolarization
As shown in Figs. 3 and 4, during long lasting (4 s) voltage clamp pulses to 0 mV in the presence of low [EGTA] in the myoplasm (1 mM), [Ca2+]SR started to decrease at the beginning of the pulse and continued to do so for the first second. After that, the [Ca2+]SR begun to rise despite that the membrane potential was kept depolarized by the voltage clamp.
Figure 5 shows simultaneous records of [Ca2+]SR and [Ca2+]myo transients elicited by pulses to 0 mV, 4 s in duration. The latter was measured with the low affinity indicator Rhod FF. Consistent with the reaccumulation of Ca2+ in the SR, the myoplasmic transient decayed during the pulse. It is reasonable to conclude that the balance between release and reuptake was shifted towards the latter and that this might reflect a decrease in SR permeability. It has been shown that, after 1 s of depolarization, the voltage sensor begins to inactivate (Brum et al. 1988b; Ríos and; Pizarro 1991). This was explored by the effect of a conditioning pulse, of the same amplitude and duration of that used to measure the [Ca2+]SR transient, on the charge movement elicited by a 100 ms voltage clamp pulse to 0 mV. This is shown in the inset of the bottom panel of Fig. 5, where charge movement currents with and without conditioning pulse are plotted. The 4 s conditioning depolarization, separated 100 ms from the test pulse, reduced the charge movement to one-third of the unconditioned value. The ensuing reduction in voltage activated permeability could easily account for the reduction of release and the net reuptake of Ca2+ by the SR. Thus, under conditions of low myoplasmic Ca2+ buffering, even very long pulses were not able to completely deplete the SR. Therefore, to achieve a more complete depletion it was necessary to add more Ca2+ buffer to the myoplasm. As shown in Figs. 3 and 4, when this was done, the free resting [Ca2+]SR decreased to a much lower level. This depleting effect of the added Ca2+ buffers happened in fibers at rest and it was augmented if the cells were stimulated. Even when starting at low [Ca2+]SR and in the presence of 40 mM EGTA in the myoplasm, pulses to 0 mV and 8 s in duration were not able to fully deplete the SR. Even when a steady level was reached, the [Ca2+]SR transients immediately changed slope and Ca2+ began to reaccumulate in the SR upon turning off the pulse. Therefore, the remnant [Ca2+]SR was enough to maintain some release. The experiment in Fig. 5 also emphasizes that, to study the control of the permeability by luminal Ca2+, shorter pulses (to avoid time and voltage dependent-inactivation of ECC) have to be used.

In a, b and c an experiment in which simultaneous intra SR and myoplasmic [Ca2+] transients were measured, with an internal solution with 1 mM EGTA, is shown. a shows the voltage clamp pulse to 0 mV. In b the [Ca2+]SR record and c the myoplasmic [Ca2+] record are shown. The latter was measured with the low affinity indicator Rhod FF. Consistent with the reaccumulation of Ca2+ in the SR, the myoplasmic transient decayed during the pulse. In Fig. 5c, d an experiment in another cell, also with a 1 mM EGTA internal solution, is shown. c is the voltage clamp pulse. d is the [Ca2+]SR. The inset shows the effect of a conditioning pulse, of the same amplitude and duration of that used to measure the intra SR transient, on the charge movement elicited by a 100 ms voltage clamp pulse to 0 mV. Charge movement currents with and without conditioning pulse are plotted, records 2 (6.1 nC/μF) and 1 (16.4 nC/μF) respectively. The 4.5 s conditioning depolarization reduced the charge movement to one-third of the unconditioned value. This would produce a reduction in voltage activated permeability that could easily account for the reduction of release and the net reuptake of Ca2+. Fiber in A and B 080415, [Rhod FF] was 178 μM in the moment when the record was taken. In c, fiber 110,515, linear capacitance was 6.1 nF
As the addition of extra buffer to the myoplasm produced in all cases the decay of [Ca2+]SR, we studied the dependence of Ca2+ release on [Ca2+]SR. This study is presented in the following section.
Measurement of SR Ca2+ permeability at different [Ca2+]SR
To find a compromise between the increased myoplasmic Ca2+ buffer power and the preservation of [Ca2+]SR, we used 5 mM EGTA in the intracellular solution. A series of experiments under this condition were conducted by simultaneously measuring [Ca2+]myo and [Ca2+]SR. With sporadic stimulation, [Ca2+]SR remained at values close to that measured at the beginning of the experiment. If pulses of moderate duration were used, 300–400 ms, the decrease in [Ca2+]SR during the pulse was far from complete. Therefore to explore the full range of [Ca2+]SR, we had to stimulate the cell with a train of pulses close enough in time to deplete the SR but not to inactivate charge movement. The outcome of such an experiment is shown in Fig. 6. Records of [Ca2+]SR, the corresponding [Ca2+]myo, release flux and permeability elicited in response to pulses to 0 mV are shown. Between the records labeled 1 and 2, two series of twenty pulses of 200 ms to 0 mV, separated by 550 ms, were applied. In the pulses during the train no optical signals were acquired because the fiber was not illuminated to avoid dye bleaching. A test pulse after each train showed a reduced initial [Ca2+] SR, thus allowing the study of the effect of this variable on the voltage activated permeability with preserved charge movement. [Ca2+]SR partially recovered after each depleting train. Figure 6d shows the release flux records obtained from the corresponding [Ca2+]myo transients in 6c. The inset in Fig. 6d shows the post pulse decay of the [Ca2+]myo transient (in grey traces) with an expanded time scale. The smooth lines superimposed to the decay of [Ca2+]myo are calculated with the best fit parameters of the removal model, assuming that release flux was null. These parameters are given in the figure legend. The Ca2+ permeability of the SR was calculated as the ratio of the release flux waveform and the corresponding [Ca2+]SR transient. The ratio is proportional to the actual permeability under the assumption that there is a linear relationship between the flux through the release pathways and the driving force. The proportionality factor is the ratio between the surface of the SR and the volume of the myoplasm. The linearity requires that the release channels are operating far from saturation. The driving force for release is taken as [Ca2+]SR, which is always much higher than the myoplasmic [Ca2+]. It is also assumed that the SR membrane voltage is null, there is likely to be little SR membrane potential added to the chemical driving force for SR Ca2+ release since the counterion flux through the SR K+ channels (Labarca and Miller 1981; Cukierman et al. 1985) or the RyRs (Gillespie and Fill 2008) would balance the charge carried by Ca2+ at any time. The permeability transient obtained gave the time course of the open probability of the RyR channels during the voltage clamp pulse.

Dependence of Ca2+ permeability on [Ca2+]SR. In a the voltage clamp pulse is shown. In b records of [Ca2+]SR before (1) and after (2) a train of depleting pulses (see text for a full explanation), in c the corresponding [Ca2+]myo and in d the release fluxes calculated from them are shown. The inset in b shows the charge movement currents measured with a short pulse applied right before the longer pulses used to measure SR depletion. The inset in d shows the decay of the [Ca2+]myo transients in b at an expanded scale, in grey. The smooth black lines superimposed to the post pulse decay of the transient are the [Ca2+] calculated with the best fit parameters of the removal model. These parameters were used to calculate the release flux and are given below. In e the permeability records, obtained as the ratio of release flux waveforms and the corresponding [Ca2+]SR transients, are shown. The [Ca2+]SR transients were smoothed (1%) before calculating the ratio in order to reduce the noise in the permeability record. The inset in e shows the same permeability records at an expanded time scale. The arrowhead indicates the beginning of the voltage clamp pulse. The vertical dashed line indicates 10 ms into the pulse, note the important difference in the activation of permeability between the high and low [Ca2+]SR condition. Fiber 211,016. Diameter: 96 μm, optical path: 84 μm,[Rhod2] was 120 μM for record 1 A and 280 μM for record 1B. The charge movement was 18.3 and 17.9 nC/μF (corresponding to records 1 and 2 respectively), Linear capacitance went from 5.5 to 5.7 nF. Parameters for release calculation: [EGTA] = 5 mM, konEGTA = 3/μM/s, koff EGTA = 5/s, [Parvalbumin] was 1 mM for record 2A and 0.3 mM for record 2B, pump rate = 1000 μM/s. Other parameters are the same as in Brum et al. (1988a)
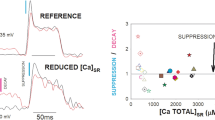
Comparison of permeability at two initial [Ca2+]SR values of 380 and 150 μM shows that the peak was moderately reduced at the lower [Ca2+]SR (Fig. 6e).
The effect of reduced [Ca2+]SR was more noticeable at early times during the pulse. The inset of Fig. 6e shows the time course of the activation of SR Ca2+ permeability with an expanded time scale. At 10 ms after the beginning of the pulse (indicated by the vertical dashed line), while the same amount of charge movement occurred in both cases, the activation of permeability was drastically less in the low [Ca2+]SR condition. Also these records showed a reduced rate of decay after the peak of the permeability in lower [Ca2+]SR. This is likely a consequence of reduced Ca2+ dependent inactivation. The average permeability, measured as the time integral of the permeability divided by the length of the integration time (the duration of the voltage clamp pulse), was not significantly modified.
In Table 1, we give the results from four fibers where the complete analysis was carried out successfully. In these four fibers, the peak permeability (column (3) in Table 1) went from 0.026 ± 0.005 to 0.021 ± 0.004 1/ms and the time averaged permeability (column (4) in Table 1) from 0.009 ± 0.003 to 0.010 ± 0.003 1/ms. While the peaks were significantly different (p = 0.035) the averages were not (p = 0.32). As shown in Fig. 6, the permeability in low [Ca2+]SR had also a longer rise time, with a 6 ms delay between peaks. This was observed in all the fibers in Table 1. The peak permeability occurred at 13 ± 1.8 ms into the pulse in reference conditions and 19.2 ± 2.6 ms after SR depletion. The difference between times to peak was statistically significant (p = 0.01). The decay from the peak was slowed down in all fibers. The half time of decay measured from the peak was 11 ± 3.3 ms in reference and 33.5 ± 3.8 ms in SR depleted condition, this difference was also statistically significant (p = 0.001).
The average of [Ca2+]SR at the end of the pulse (400 ms), in these four fibers, was 102 ± 26 μM in the condition of higher resting [Ca2+]SR. When starting from lower resting [Ca2+]SR, the average [Ca2+]SR at 400 ms was 41 ± 10 μM. Despite that one is 40% of the other, these values were not significantly different (p = 0.15). The permeability at 400 ms was 0.006 + 0.002 and 0.007 + 0.003 ms− 1, in high and low resting [Ca2+]SR respectively, also not significantly different.
A comparison of the effect of high myoplasmic [EGTA] and [BAPTA] on the SR Ca2+ permeability
The application of high [EGTA] to the myoplasm from the cut ends had the effect of decreasing [Ca2+]SR. This was not specific of EGTA since it also occurred with BAPTA. It was probably due to the capture of the Ca2+ that leaked at rest from the SR by the extrinsic myoplasmic buffer. As a consequence, by the time we restarted to apply pulses to the cell to measure Ca2+ release, the [Ca2+]SR was drastically reduced. Based on the previous data, we think that it is reasonable to conclude that if we confine the study to cells where the [Ca2+]SR in the presence of high myoplasmic buffer fell within the same narrow range, differences in Δ[Ca2+]SR in the presence of different buffers in the myoplasm would reflect differences in the permeability. We compared the effect of EGTA and BAPTA at similar equilibrium buffer power. Since at pH 7.0 kdBAPTA = 0.2 μM and kdEGTA = 0.4 μM, we used twice the concentration of EGTA. Under these conditions and assuming that BAPTA binds Ca2+ 100 times faster the EGTA, the length travelled by Ca2+ before being captured is 7 times smaller for BAPTA (see Discussion). This is a measure of the ability of the extrinsic buffer to confine the Ca2+ signal and therefore to interfere with recruitment of neighboring channels by CICR.
Figure 7 shows records of [Ca2+]SR transients from two cells, each in the starting reference condition (5 mM EGTA intracellular solution) and after the application of 40 mM EGTA (panel A) or 20 mM BAPTA (panel B). In these cells, and the others included in this comparison [Ca2+]myo was not measured. In both high buffer conditions, the resting [Ca2+]SR was strongly reduced compared to that measured at the beginning of the experiments. The rate of change of the [Ca2+]SR transient in the presence of 20 mM BAPTA was clearly smaller than in 40 mM EGTA. As a consequence, the Δ[Ca2+]SR at 200 ms into the pulse was also smaller in BAPTA. The insets show the charge movement measured during 100 ms pulses to 0 mV applied immediately before the long pulses used to measure SR depletion.

The figure shows, in panels a and b, records of [Ca2+]SR transients from two cells, in reference conditions (5 mM EGTA intracellular solution, black thin traces in panels a and b) and in 40 mM EGTA (panel a, thick grey traces) or 20 mM BAPTA (panel b, thick black traces). In both high buffer conditions, the resting [Ca2+]SR was strongly reduced compared to that measured at the beginning of the experiments. The [Ca2+]SR transient in the presence of 20 mM BAPTA was clearly smaller than in 40 mM EGTA. The arrow heads indicate the beginning and the end of the voltage clamp pulses to 0 mV. The vertical dashed lines indicate 200 ms after the beginning of the pulse, where the amplitudes of the transients were measured. The insets show the charge movement measured during 100 ms pulses to 0 mV applied immediately before the long pulses used to measure SR depletion. Fiber identifier in A: 180,315. In this cell, the charge movement was 29.8 nC/μF in reference and 29.4 nC/μF in high EGTA. The records are shown in the inset with the same trace code as the [Ca2+]SR transients. In the same period linear capacitance went from 9.3 to 9.4 nF. Fiber identifier in B: 050715b. The charge movement was 18.4 nC/μF in reference and 18.1 nC/μF in high BAPTA. The records are shown in the inset with the same trace code as the [Ca2+]SR transients. The linear capacitance went from 8.3 to 8.6 nF. In c, d the average results from four cells in EGTA and five cells in BAPTA are compared. Only Δ[Ca2+]SR (measured 200 ms after the beginning of the pulse) was significantly smaller in 20 mM BAPTA compared to 40 mM EGTA. In the experiments with EGTA, the average charge movement before the application of the buffer was 21.6 ± 4.3 nC/μF and 20.8 ± 4.1 nC/μF. The time elapsed since the application of the buffer was 36 ± 9.5 min. In the experiments with BAPTA, the average charge movement before the application of the buffer was 23.2 ± 7.1 nC/μF and 22.4 ± 6.3 nC/μF. The time elapsed since the application of the buffer was 39.8 ± 4.6 min. In e the average of time constants obtained in single exponential fits to the transients in 40 mM EGTA (grey) and 20 mM BAPTA (black) are plotted. The two averages are significantly different
Figure 7 also shows the average results from four cells in 40 mM EGTA and five cells in 20 mM BAPTA, panels c and d respectively. The comparison was carried out in cells where the effect of the buffer was measured at similar resting [Ca2+]SR and with conserved charge movement. As a consequence, resting [Ca2+]SR in high buffer were not significantly different. In 20 mM BAPTA, the resting [Ca2+]SR = 88.8 ± 13.6μM (n = 5) and in EGTA the resting [Ca2+]SR = 98.2 ± 15.6μM (n = 4). The same applies to the charge moved during a 100 ms pulse to 0 mV before and after the application of the high buffer internal solution. The average change in [Ca2+]SR measured at 200 ms into the pulse was significantly smaller, roughly one-half, in BAPTA compared to EGTA, 30.2 ± 3.5 μM (n = 5) and 72.2 ± 8.6 mM (n = 4) respectively (p = 0.004, unpaired t-test).
Another series of experiments was carried out using lower concentrations of both buffers in order to attain an intermediate resting [Ca2+]SR. The solutions were designed with the same criteria as above, i.e. to have the same equilibrium buffer power. With this purpose, internal solutions of 10 mM BAPTA or 20 mM EGTA, both with 20 nM free [Ca2+], were used in this experimental series. After equilibrating the myoplasm with the solution in the lateral pools, fibers whose resting free [Ca2+]SR values were in the vicinity of 200 μM were included in the study. The other a requirement for a cell to be ncluded was that the charge movement was conserved. Figure 8 shows records of + Δ[Ca2+]SR from two experiments, one in 20 mM EGTA (Fig. 8a) and the other in 10 mM BAPTA (Fig. 8c). The corresponding charge movement currents are shown in the insets. Figure 8b, d shows the average results, for 20 mM EGTA and 10 mM BAPTA, respectively. The resting free [Ca2+]SR was not statistically different, 201 ± 6 μM (n = 6) in 10 mM BAPTA and 221 ± 13 μM (n = 4) in 20 mM EGTA. The amplitude of Δ[Ca2+]SR signal, measured at 200 ms into a pulse to 0 mV, was 47 ± 9 μM in 10 mM BAPTA and 107 ± 24 μM in 20 mM EGTA (statistically different, p = 0.046). Thus, 10 mM BAPTA suppressed (compared to fibres in 20 mM EGTA) the amplitude of Δ[Ca2+]SR approximately in the same proportion as 20 mM BAPTA (compared to 40 mM EGTA).

Records of [Ca2+]SR transients from two cells in reference conditions (5 mM EGTA intracellular solution, thin black traces) and in 20 mM EGTA (panel a, thick grey traces) or 10 mM BAPTA (c, thick black traces) are shown. The arrow heads indicate the beginning and the end of the voltage clamp pulses to 0 mV. The vertical dashed lines indicate 200 ms into the pulse, where the amplitudes of the transients were measured. The insets show the charge movement measured during 100 ms pulses to 0 mV applied immediately before the long pulses used to measure SR depletion, with the same trace code as the [Ca2+]SR transients. Fiber in A: 041217, charge movement was 20.6 and 19.4 nC/μF, in reference and 20 mM EGTA, respectively. In the same period linear capacitance went from 6.4 to 6.9 nF. Fiber in B: 220,318. Charge movement was 29.3 and 30 nC/μF, in reference and 10 mM BAPTA, respectively. The linear capacitance went from 5.7 to 5.6 nF. In b and d the average results from four cells in EGTA and six cells in BAPTA are compared, only Δ[Ca2+]SR (at 200 ms after the beginning of the pulse) was significantly smaller in 10 mM BAPTA compared to 20 mM EGTA. In the experiments with EGTA, the average charge movement before the application of the buffer was 25.4 ± 2.8 and 23.9 ± 2.8 nC/μF. The time elapsed since the application of the buffer was 29.5 ± 2.5 min. In the experiments with BAPTA, the average charge movement before the application of the buffer was 24.4 ± 2.2 nC/μF and 23.8 ± 3.5 nC/μF. The time elapsed since the application of the buffer was 36.3 ± 3.4 min. In e the average of time constants obtained in single exponential fits to the transients in 20 mM EGTA (grey) and 10 mM BAPTA (black) are plotted. The two averages are significantly different
The time course of the [Ca2+]SR transients in high buffer was roughly exponential. To further quantify the effect of the buffers on the SR permeability, decaying exponential functions (plus a positive constant) were fitted to the transient. The time constants in 40 mM EGTA had an average of 148.7 + 19.4 ms. In 20 mM BAPTA the average time constant was 409.9 + 42.3 ms. The difference between means was statistically significant (p = 0.001, Fig. 7e). In 20 mM EGTA the time constant was 197.2 + 31.4 ms, while in 10 mM BAPTA the average was 392.2 + 24.7 ms. Again, these means were statistically different (p = 0.01, Fig. 8e). When comparing the time constants in the two concentrations of EGTA used, that correspond to two different starting values of [Ca2+]SR, the averages were not statistically different. The same happens for the two concentrations of BAPTA. The slower kinetics in BAPTA explains the observed difference in amplitudes measured at 200 ms into the pulse.
This supports that in the presence of BAPTA containing internal solutions, with roughly the same driving force for release and with the voltage sensing mechanism unaltered, the Ca2+ permeability of the SR is reduced when compared to that in EGTA internal solutions.
Discussion
One main observation in this study is that the myoplasmic application of 10 or 20 mM BAPTA suppressed the amplitude and slowed down the rate of change of the [Ca2+]SR transient when compared to 20 or 40 mM EGTA respectively.
A second main observation is that the time averaged Ca2+ permeability of the SR was not changed when resting [Ca2+]SR was reduced from the initial value of about 400 μM to much lower levels, in the range of 100 μM (Table 1). In spite of this, important kinetic differences were observed.
The effect of BAPTA supports the existence of a CICR component in the overall depolarization elicited Ca2+ release. The change in kinetics of the permeability time course could also be interpreted as a modification of CICR. Although the interpretation of these two findings is interrelated, in what follows the two effects will be discussed separately and then compared to work by other laboratories.
The dependence of SR Ca2+ permeability on [Ca2+]SR
The effect of [Ca2+]SR was studied by depleting the reservoir with a train of pulses, as explained in Results. This is documented in Fig. 6 and Table 1. The reduction of the [Ca2+]SR had the trivial effect of reducing the amplitude of the Ca2+ release flux, calculated from the myoplasmic [Ca2+] transient as explained in Methods. Not only was the peak release drastically reduced but the rate of decay after the peak was also slowed down. This has been previously observed and interpreted as a reduction in the rate of Ca2+ dependent inactivation (Schneider and Simon 1988; Pizarro and Ríos 2004; Olivera and Pizarro 2010).
The calculated permeability signal showed that the peak value was slightly reduced and that the rate of decay after the peak was also reduced (Fig. 6E). The ratio between peak and quasi stationary permeability was about two, slightly higher in higher [Ca2+]SR.
The activation kinetics of the permeability was also slowed down. At early times during the pulse the effect of reduced [Ca2+]SR was quite pronounced. For instance, in the inset of Fig. 6E, at 10 ms into the pulse the permeability was 3 times bigger at high [Ca2+]SR. At this time, 75% of the charge has moved in both conditions, therefore the difference was not due to changes in voltage activation. Thus, the delayed peak of the permeability time course could not be due only to a slowed down Ca2+ dependent inactivation or to a reduction in the speed of activation.
These findings are not consistent with a purely voltage dependent release with fast Ca2+ dependent inactivation. In this case, a potentiation of permeability is expected, as a consequence of the reduced inactivation due to the lower [Ca2+]SR that, in turn, would produce a smaller [Ca2+] at the inactivation site on the myoplasmic side of the RyRs.
CSQ-mediated regulation by luminal [Ca2+]SR, as described in heart muscle (Gyorke et al. 2002) and in mammalian skeletal muscle (Sztretye et al. 2011b), could also account for the reduction in permeability at early times during the pulse. The observation that the quasi stationary level that followed the peak did not show an obvious tendency to decline, even as [Ca2+]SR got to very low values, suggests that there is no luminal regulation by [Ca2+]SR.
A dual mechanism, consisting in a voltage dependent Ca2+ release that secondarily triggers CICR, is also consistent with these observations.
Reducing [Ca2+]SR and the consequent reduction of release will also reduce the rate of activation of the CICR component, contributing to the delay in the peak permeability (Fig. 6e inset). As the CICR component also inactivates by Ca2+, the reduced release will slow down this process, allowing CICR to proceed for a longer time. Thus, the CICR component of release is expected to be reduced, although not necessarily abolished by low [Ca2+]SR.
The RyRαs, the likely pathways for voltage dependent release, show Ca2+ dependent inactivation, either when reconstituted in bilayers or in isolated TC vesicles (Murayama and Kurebayashi 2011). The release flux in mammals decays after the peak with a time course that resembles that of the amphibian (Shirokova et al. 1996), suggesting similar kinetics of Ca2+ dependent inactivation in both species. Sztretye et al. (2011b) reported that 10 mM BAPTA doubled the peak of the permeability, compared to the value found in 10 mM EGTA. They attributed this effect to the inhibition of Ca2+ dependent inactivation. In spite of this, their permeability waveform showed a marked fast decay after the peak, that they attributed to luminal regulation. If this interpretation is correct it appears that the Ca2+ dependent inactivation is a very fast process, which has already reached a steady state by the time of the peak, and that the decay in permeability that follows is due to the reduction in [Ca2+]SR.
In frog skeletal muscle fibers, Pape and Carrier (1998) showed that the permeability elicited by very small depolarization, to − 60 mV or more negative, increased when the [Ca2+] in the SR was reduced. They interpreted this observation as a reduction of Ca2+ dependent inactivation on the same channel through which Ca2+ was released. The time course of their estimated permeability did not show any decay during the pulse up to 300 ms. Thus, even in the presence of Ca2+ dependent inactivation, a rather steady time course is possible. This is consistent with the time course of the precursor release observed by Shirokova and Ríos (1997).
In any case, the lower [Ca2+]SR is expected to increase the voltage activated permeability.
All of the above accounts for the fact that the time averaged permeability was unchanged upon reduction of [Ca2+]SR. The reduction in the rate of inactivation compensated the reduced peak and reduced rate of activation, as shown in the inset of Fig. 6d.
The effect of high myoplasmic [BAPTA] on SR Ca2+ permeability
As the reduction of [Ca2+]SR was an unavoidable consequence of the application of high [BAPTA] or [EGTA] to the internal solution, we restricted the study of the effect of the myoplasmic application of extrinsic buffers to fibers that upon the application of the buffers had the resting [Ca2+]SR within a narrow range. Starting at the same [Ca2+]SR the intra SR buffering would operate with similar buffer power. Furthermore as [Ca2+]SR was low and likely much below the kd of CSQ (Pape et al. 2007), then Δ[Ca2+]SR was likely proportional to Δ[Ca]Total. Also this condition gave roughly the same driving force for release. As a consequence, the amplitude of Δ[Ca2+]SR in a given time (200 ms) would measure the average flux, and in turn would be proportional to the average permeability. Even if these conditions were not exactly met the difference between EGTA and BAPTA is best interpreted as a reduction in SR Ca2+ permeability by BAPTA.
This conclusion is supported by the effect of the added buffers on the time constant of the Δ[Ca2+]SR transients. As mentioned in results, they had an approximately exponential time course. The time constants in BAPTA roughly double those in EGTA. On the other hand, the time constants in each buffer are similar at the two concentrations tested. The simplest interpretation of an exponential relaxation is that of a two compartments system equilibrating through a barrier of constant permeability. Within this framework, the time constant is inversely proportional to the permeability. Therefore, the doubling of the time constant reflects the reduction of the permeability to one-half. This analysis does not necessarily imply that the time course of permeability lacks a peak, but that the equilibration between the compartments is dominated by the rather steady permeability attained after the peak. The kinetic effect is consistent with our interpretation that the fast buffer BAPTA reduced the permeability when compared with the slower EGTA. We cannot affirm that in 20 or 40 mM the permeability is not modified respect to the more physiological condition in 5 mM EGTA. The same time constant value in 10 and 20 mM BAPTA supports the idea that the effect due to buffering is maximal.
The difference in Ca2+ buffering between chelators with similar kd was shown to originate in the different on rates, the faster buffers being more effective to capture Ca2+ as it diffuses away from the source. This is characterized by an average distance before capture (Neher 1986; Stern 1992; Pape et al. 1995) given by
where KonB is the on rate of the buffer, CB is the buffer concentration and DCa is the Ca2+ diffusion coefficient. Given the ranges of estimates of DCa − 220 (Naraghi and Neher 1997) to 300 μm2/s (Pape et al. 1995) and of on rates for the two chelators -KonBAPTA between 1 and 3 μM−1 s− 1 (Harafuji and Ogawa 1980) and KonEGTA between 100 (Kits et al. 1999) to 400 μM−1 s−1(Naraghi and Neher 1997)- a range of λ in 20 mM BAPTA and 40 mM EGTA was calculated with Eq. 3a. λBAPTA was between 5 and 12 nm while λEGTA was between 43 and 87 nm.
The [Ca2+] as a function of the distance r from the point source is well approximated by (Neher 1986; Stern 1992; Pape et al. 1995)
where ϕ is source intensity and Cao is the resting [Ca2+] .
The RyRβs are the likely candidates to be Ca2+ activated. The center to center distance between the nearest RyRβ in the parajunctional clusters and the closest RyRα in the double row array at the junctional TC is 28 nm (Felder and Franzini-Armstong 2002). Therefore BAPTA could successfully antagonize Ca2+ dependent activation while EGTA would be ineffective.
Using Eq. 3b, the [Ca2+] at 28 nm from a point source of the same intensity in 40 mM EGTA is about 70% that in the presence of 5 mM EGTA.
On the other extreme of our experimental interventions, the value of λ in 20 mM BAPTA and [Ca2+]SR = 100 μM is 71% the one in 10 mM BAPTA and [Ca2+]SR = 200 μM. Assuming that the source intensity is linearly related to [Ca2+]SR, the [Ca2+] at 28 nm from the source in 20 mM BAPTA is 29% that in 10 mM BAPTA. This important reduction in [Ca2+] did not further suppress the Δ[Ca2+]SR, suggesting that the effect of 10 mM BAPTA on SR Ca2+ permeability was maximal (Figs. 7, 8).
More important is the comparison between [Ca2+] at 28 nm from the source with 40 mM EGTA and 10 mM BAPTA, in this range is where the maximum suppressive effect of BAPTA occurred (Figs. 7, 8). In 40 mM EGTA, the [Ca2+] is 3 times bigger than in 10 mM BAPTA.
All of the above is an oversimplification, with pulses to 0 mV many sources would be simultaneously open and the contributions from individual channels would add up, especially in EGTA. If this situation is considered, the buffering effect is even clearer. With pulses to 0 mV, the charge movement is about 80% of the maximum. The voltage dependent activation of release follows a power function of the charge movement. The exponent is between 2 and 4 (Ríos et al. 1993; Simon and Hill 1992). Therefore, the expected activation would be between 41 and 64% of the maximum. Assuming 50% activation, i.e. every other channel in the junctional double row is open at any given time during the pulse, the [Ca2+] along the line of center of the first row of parajunctional channels could be calculated using Eq. 3b for each source and adding the individual contributions. In the case of the upper estimates of λ (with the highest DCa and the slowest konB for each buffer), the comparison between 40 mM EGTA and 10 mM BAPTA shows that, in the former condition, the [Ca2+] is 14 times bigger.
The permeability suppressed by BAPTA is likely due to inhibition of CICR as a consequence of Ca2+ buffering. Based on our data, the reduction in the permeability cannot be exactly quantified but seems reasonable to tentatively conclude that the treatment suppressed 50% of the total permeability.
Given that the amount of RyRα and RyRβ are similar (Felder and Franzini-Armstrong 2002) and that currents through frog channels reconstituted in lipid bilayers show two modes (maybe due to α and β) that only differ by 30% (Kettlun et al. 2003), it seems reasonable to postulate that the contribution to the total release by each type is about the same. This is consistent with the rough estimate based on our data if CICR results in the activation of one additional channel on average.
Although a pharmacological effect of BAPTA cannot be entirely excluded, the lack of effect of 8 mM BAPTA on Ca2+ release in response to pulses to − 60 mV reported by Pape et al. (2002) argue against it.
Comparison with previous work
A study by Fénelon et al. (2012) found that, during a voltage clamp pulse to − 20 mV, as the [Ca2+]SR went down so did the SR Ca2+ permeability. Their study was based on cytoplasmic measurements of Ca2+ release flux and an estimate of [Ca2+]SR using the total Ca released and their parameters for CSQ obtained by fitting a Hill equation to a different set of data. The permeability estimated by Fénelon et al. (2012) declined from the peak in two stages. First, it declined to a level approximately 50% of the peak. This level was rather steady until 70 ms and the permeability continued to decline towards zero. This is not entirely consistent with our findings, as we found a quasi steady permeability after the decline from the peak, lasting 400 ms. It is possible that they have more complete depletion of the SR due to their higher release flux and that this might be the reason for their second stage of decline.
Pape et al. (2002) studied the effect of SR Ca on release at − 45 mV. Their Fig. 6 shows release wave forms that are quite similar to ours at 0 mV. In the SR depleted case the rate of rise of release was slowed down, its peak was delayed and the decline after the peak was also slowed down. In the original paper (Pape et al. 2002) they corrected the release flux by the total Ca in the SR and reported an increase in permeability by a factor of two compared to the fully loaded case. When they correct for [Ca2+]SR (Fenelon et al. 2012, Fig. 4d), the newly calculated permeability is now 60% of the fully loaded case, making their permeability analysis at − 45 mV quite similar to ours at 0 mV.
There are quantitative differences between our results (Fig. 6) and the above mentioned findings of Fenelon et al. (2012). The suppression in the case of Fenelon et al. (2012) is stronger than in this study. A more important contribution of CICR at the less depolarized voltages that they explored (− 45 and − 20 mV) compared to ours (0 mV) might have contributed to the difference. There are some lines of evidence in favor of this interpretation. Ca2+ sparks, thought to be through RyRβ activated by CICR, made up most of the release at mild depolarizations (Shirokova and Rios 1997). A maximum in the peak to steady release ratio is characteristic of frog muscle and absent in mammalian muscle (Shirokova et al. 1996). It is probably due to activation of RyRβ in the frog because it appears in mammalian cells when they are transfected with RyR3 (Pouvreau et al. 2007; Ríos 2018). Therefore, it is in all likelihood an indication of CICR and it is placed at moderately depolarized voltages, about − 40 mV.
Our findings regarding the effect of BAPTA are broadly consistent with those of Jacquemond et al. (1991), Csernoch et al. (1993) and Fenelon and Pape (2002), although our method cannot detect differential effects on peak and quasi steady components of the SR Ca2+ permeability. Most likely, both the peak and the quasi steady level were suppressed, as it is difficult to account for a reduction in 50% of the Ca2+ released in 200 ms at the expense of the early peak alone.
We favor the idea that CICR occurs via RyRβs, mostly based in the work by Murayama and Ogawa (2001) that showed that CICR is inhibited in the RyRαs when they are located in the SR. This inhibition is independent of the interaction with the DHPR. In spite of this, CICR mediated by RyRα cannot be entirely ruled out based on the data presented here. The distance between the nearest RyRα in the double row array is basically the same as the distance between the nearest RyRα and RyRβ (Felder and Franzini-Armstrong 2002). Therefore, 10 mM BAPTA or higher should be equally effective to antagonize their activation, if these channels could be activated by Ca2+. The CICR within the double row array might be via those channels not facing DHPRs. Also a self regulatory process, in which the Ca2+ coming through the pore increased the open time of a channel that was already opened by the voltage sensor (Tripathi and Meissner 1996; Pape and Carrier 1998) cannot be excluded, although this kind of CICR should be less sensitive to high [BAPTA] (Pape et al. 2002).
From the data presented in this study, the emerging picture is that of a highly Ca2+ sensitive, highly regenerative CICR process that makes a sizable contribution to the activation of Ca2+ release. The size of this contribution is about 50% of the total release, at least in [Ca2+]SR ≤ 200 μM.
These conclusions differ from those drawn from a study of expression of both frog isoforms in 1B5 skeletal muscle mammalian cells (Kashiyama et al. 2010). When α and β were coexpressed the voltage triggered Ca2+ signal was not statistically different from that obtained by expressing α alone. Furthermore, in the cells that coexpressed α and β the treatment with procaine, a blocker of caffeine triggered β mediated Ca 2+ release, did not suppress the voltage dependent Ca2+ transient. Their conclusion was that the role of β isoform and CICR in the physiological release was null. In order to be conclusive this study would have needed morphological data showing that the location of the expressed β was at the same distance from the α as in the native systems.
Another recent study also questioned an important role of RyRβ, and consequently of CICR, in zebra fish skeletal muscle. The skeletal muscle cells from a RyRβ knockout lacked the parajunctional channels and did not show Ca2+ sparks. The RyRb animal had the same performance in swimming tests as the wild type (Perni et al. 2015). Therefore they concluded that the RyRβs were not physiologically relevant. They proposed that during the massive release through RyRαs during an action potential the parajunctional RyRβs got inactivated by Ca2+. Only after the action potential finished and voltage dependent release stopped, the RyRβs might activate by the residual Ca2+ and prolong Ca 2+release in non mammalian species. In a voltage clamp experiment, this CICR component would contribute only to the post peak, lower, quasi steady level. Although this interpretation is not entirely inconsistent with our data, it seems hard to understand how the reduction in [Ca2+]SR does not potentiate the peak permeability as a consequence of reduced inactivation. Therefore, it seems more reasonable that CICR contributes to both the peak and steady levels.
References
Beard NA, Laver DR, Dulhunty AF (2004) Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol 85(1):33–69
Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C (1988) Structural evidences for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol 107:2587–2600
Brum G, Ríos E, Stefani E (1988a) Effects of extracellular calcium on calcium movements of excitation-contraction coupling in frog skeletal muscle fibres. J Physiol 398:441–473
Brum G, Fitts R, Pizarro G, Ríos E (1988b) Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation-contraction coupling. J Physiol 398:475–505
Canato M, Scorzeto M, Giacomello M, Protasi F, Reggiani C, Stienen GJ (2010) Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc Natl Acad Sci USA 107:22326–22331
Cheng H, Lederer WJ (2008) Calcium sparks. Physiol Rev 88(4):1491–1545
Csernoch L, Jacquemond V, Schneider MF (1993) Microinjection of strong calcium buffers suppresses the peak of calcium release during depolarization in frog skeletal muscle fibers. J Gen Physiol 101(2):297–333
Cukierman S, Yellen G, Miller C (1985) The K+ channel of sarcoplasmic reticulum. A new look at Cs + block. Biophys J 48(3):477–484
De Armas R, González S, Brum G, Pizarro G (1998) Effects of 2,3-butanedione monoxime on excitation-contraction coupling in frog twitch fibres. J Mus Res Cell Motil 19:961–977
Donoso P, Prieto H, Hidalgo C (1995) Luminal calcium regulates calcium release in triads isolated from frog and rabbit skeletal muscle. Biophys J 68:507–515
Endo M (2009) Calcium-induced calcium release in skeletal muscle. Physiol Rev 89(4):1153–1176
Endo M, Tanaka M, Ogawa Y (1970) Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature 228:34–36
Felder E, Franzini-Armstrong C (2002) Type 3 ryanodine receptors of skeletal muscle are segregated in a parajunctional position. Proc Natl Acad Sci USA 99(3):1695–1700
Fénelon K, Pape PC (2002) Recruitment of Ca2+ release channels by calcium-induced Ca2+ release does not appear to occur in isolated Ca2+ release sites. J Physiol 544(3):777–791
Fénelon K, Lamboley CR, Carrier N, Pape PC (2012) Calcium buffering properties of sarcoplasmic reticulum and calcium-induced Ca2+ release during the quasi-steady level of release in twitch fibers from frog skeletal muscle. J Gen Physiol 140(4):403–419
Fill M, Copello J (2002)). Ryanodine receptors calcium channels. Physiol Rev 82:893–922
Gillespie D, Fill M (2008) Intracellular calcium release channels mediate their own countercurrent: the ryanodine receptor case study. Biophys J 95(8):3706–3714
González A, Ríos E (1993) Perchlorate enhances transmission in skeletal muscle excitation contraction coupling. J Gen Physiol 102:373 421
Györke S, Györke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF (2002) Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci 1(7):1454–1463
Harafuji H, Ogawa Y (1980) Re-examination of the apparent binding constant of ethylene glycol bis(beta-aminoethyl ether)-N,N,N′,N′-tetraacetic acid with calcium around neutral pH. J Biochem 87(5):1305–1312
Hollingworth S, Gee KR, Baylor SM (2009) Low-affinity Ca indicators compared in measurements of skeletal muscle Ca2+ transients. Biophys J 97(7):1864–1872
Jacquemond V, Csernoch L, Klein MG, Schneider MF (1991) Voltage-gated and calcium-gated calcium release during depolarization of skeletal muscle fibers. Biophys J 60(4):867–873
Jong DS, Pape PC, Chandler WK, Baylor SM (1993) Reduction of calcium inactivation of sarcoplasmic reticulum calcium release by fura-2 in voltage-clamped cut twitch fibers from frog muscle. J Gen Physiol 102:333–370
Jong DS, Pape PC, Baylor SM, Chandler WK (1995) Calcium inactivation of calcium release in frog cut muscle fibers that contain millimolar EGTA or Fura-2. J Gen Physiol 106:337–388
Kabbara AA, Allen DG (2001) The use of the indicator fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J Physiol 534:87–97
Kashiyama T, Murayama T, Suzuki E, Allen PD, Ogawa Y (2010) Frog alpha- and beta-ryanodine receptors provide distinct intracellular Ca2 + signals in a myogenic cell line. PLoS One 12;5(7):e11526
Kettlun C, González A, Ríos E, Fill M (2003) Unitary Ca2 + current through mammalian cardiac and amphibian skeletal muscle ryanodine receptor channels under near-physiological ionic conditions. J Gen Physiol 122(4):407–417
Kits KS, de Vlieger TA, Kooi BW, Mansvelder HD (1999) Diffusion barriers limit the effect of mobile calcium buffers on exocytosis of large dense cored vesicles. Biophys J. 76(3):1693–1705
Kovacs L, Ríos E, Schneider MF (1983) Measurement and modification of free calcium transients in frog skeletal muscle fibres by metallochromic indicator dye. J Physiol 343:161–196
Labarca PP, Miller C (1981) A K+-selective, three-state channel from fragmented sarcoplasmic reticulum of frog leg muscle. J Membr Biol 61(1):31–38
Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Ríos E (2006) Depletion “skraps” and dynamic buffering inside the cellular calcium store. Proc Natl. Acad Sci USA 103:2982–2987
Manno C, Figueroa LC, Gillespie D, Fitts R, Kang C, Franzini-Armstrong C, Rios E (2017) Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc Natl Acad Sci USA 114(4):E638–E647
Melzer W, Ríos E, Schneider MF (1987) A general procedure for determining calcium release in skeletal muscle fibers. Biophys J 51:849–864
Murayama T, Kurebayashi N (2011) Two ryanodine receptor isoforms in nonmammalian vertebrate skeletal muscle: possible roles in excitation-contraction coupling and other processes. Prog Biophys Mol Biol 105(3):134–144
Murayama T, Ogawa Y (2001) Selectively suppressed Ca2+-induced Ca2 + release activity of alpha-ryanodine receptor (alpha-RyR) in frog skeletal muscle sarcoplasmic reticulum: potential distinct modes in Ca2 + release between alpha- and beta-RyR. J Biol Chem 26(4):2953–2960
Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17(18):6961–6973
Neher E (1986) Concentration profiles of intracellular calcium in the presence of a diffusible chelator. In: Heinemann U, Klee M, Neher E, Singer W (eds) Calcium electrogenesis and neuronal functioning. Springer Verlag, Berlin, pp 80–96
Olivera JF, Pizarro G (2010) A reappraisal of the Ca2+ dependence of fast inactivation of Ca2+ release in frog skeletal muscle. J Muscle Res Cell Motil 31(2):81–92
Olivera JF, Pizarro G (2016) Excitation contraction uncoupling by high intracellular [Ca2+] in frog skeletal muscle: a voltage clamp study. J Muscle Res Cell Motil 37(4–5):117–130
Pape PC, Carrier N (1998) Effect of sarcoplasmic reticulum (SR) Calciem content on SR Calcium release by small voltage-clamp depolarizations in frog cut skeletal muscle fibers equilibrated with 20 mM EGTA. J Gen Physiol 112:161–179
Pape PC, Jong DS, Chandler WK (1995) Calcium release and its voltage dependence in frog cut muscle fibers equilibrated with 20 mM EGTA. J Gen Physiol 106:259–336
Pape PC, Fénelon K, Carrier N (2002) Extra activation component of calcium release I frog muscle fibers. J Physiol 542(3):867–889
Pape PC, Fénelon K, Lamboley CR, Stachura D (2007) Role of calsequestrin evaluated from changes in free and total calcium concentrations in the sarcoplasmic reticulum of frog cut skeletal muscle fibres. J Physiol 581(Pt 1):319–367
Perni S, Marsden KC, Escobar M, Hollingworth S, Baylor SM, Franzini-Armstrong C (2015) Structural and functional properties of ryanodine receptor type 3 in zebrafish tail muscle. J Gen Physiol 145(3):173–184
Pizarro G, Ríos E (2004) How source content determines intracellular Ca2+ release kinetics. Simultaneous measurement of [Ca2+] transients and [H+] displacement in skeletal muscle. J Gen Physiol 124:239–258
Pouvreau S, Royer L, Yi J, Brum G, Meissner G, Ríos E, Zhou J (2007) Ca2+ sparks operated by membrane depolarization require isoform 3 ryanodine receptor channels in skeletal muscle. Proc Natl Acad Sci USA 104(12):5235–5240
Rios E, Pizarro G (1991) Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol Rev 71:849–908
Ríos E (2018) Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J Gen Physiol 150(4):521–537
Ríos E, Pizarro G (1988) Voltage sensors and calcium channels of excitation-contraction coupling. NIPS 3:223–227
Ríos E, Karhanek M, Ma J, González A (1993) An allosteric model of the molecular interactions of excitation-contraction coupling in skeletal muscle. J Gen Physiol 102(3):449–481
Robin G, Berthier C, Allard B (2012) Sarcoplasmic reticulum Ca2+ permeation explored from the lumen side in mdx muscle fibers under voltage control. J Gen Physiol 139:209–218
Rudolf R, Magalhães PJ, Pozzan T (2006) Direct in vivo monitoring of sarcoplasmic reticulum Ca2+ and cytosolic cAMP dynamics in mouse skeletal muscle. J Cell Biol 173:187–193
Schneider MF, Simon BJ (1988) Inactivation of calcium release from the sarcoplasmic reticulum in frog skeletal muscle. J Physiol 405:727–745
Shirokova N, Ríos E (1997) Small event Ca2+ release: a probable precursor of Ca2+ sparks in skeletal muscle. J Physiol 502(1):3–11
Shirokova N, García J, Pizarro G, Ríos E (1996) Ca2+ release from the sarcoplasmic reticulum compared in amphibian and mammalian skeletal muscle. J Gen Physiol 107:1–187
Simon BJ, Hill DA (1992) Charge movement and SR calcium release in frog skeletal muscle can be related by a Hodgkin-Huxley model with four gating particles. Biophys J 61(5):1109–1116
Stern MD (1992) Buffering of calcium in the vicinity of a channel pore. Cell Calcium 13:183–192
Stern M, Pizarro G, Ríos E (1997) Local control model of excitation contraction coupling in skeletal muscle. J Gen Physiol 110:415–440
Sutko J, Airey JA (1996) Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function? Physiol Rev 76:1027–1071
Sztretye M, Yi J, Figueroa L, Zhou J, Royer L, Ríos E (2011a) D4cpv-calsequestrin: a sensitive ratiometric biosensor targeted to the calcium store of skeletal muscle. J Gen Physiol 138:211–229
Sztretye M, Yi J, Figueroa L, Zhou J, Royer L, Allen PD, Brum G, Ríos E (2011b) Measurement of RyR permeability reveals a role of calsequestrin in termination of SR Ca2+ release in skeletal muscle. J Gen Physiol 138:231–247
Tang S, Wong HC, Wang ZM, Huang Y, Zou J, Zhuo Y, Pennati A, Gadda G, Delbono O, Yang JJ (2011) Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc Natl Acad Sci USA 108:16265–16270
Tripathy A, Meissner G (1996) Sarcoplasmic reticulum luminal Ca2+ has access to cytosolic activation and inactivation sites of skeletal muscle Ca2+ release channel. Biophys J 70:2600–2615
Ziman AP, Ward CW, Rodney GG, Lederer WJ, Bloch RJ (2010) Quantitative measurement of Ca2+ in the sarcoplasmic reticulum lumen of mammalian skeletal muscle. Biophys J 99:2705–2714
Acknowledgements
This work was funded by PEDECIBA and UdelaR.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Olivera, J.F., Pizarro, G. A study of the mechanisms of excitation–contraction coupling in frog skeletal muscle based on measurements of [Ca2+] transients inside the sarcoplasmic reticulum. J Muscle Res Cell Motil 39, 41–60 (2018). https://doi.org/10.1007/s10974-018-9497-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-018-9497-9