
Abstract
We investigated crystal structure and the local structure changes during the thermal decomposition of hydromagnesite by using in situ high-temperature XRD and ex situ high-temperature X-ray total scattering measurements. Hydromagnesite displayed anisotropic thermal expansion up to 220 °C. The a and c lattice parameters exhibited an increase trend with temperature, whereas the b lattice parameter and β angle did not show a regular trend with temperature. The relative expansion between 25 and 220 °C followed the c/c0 > a/a0 \(\gg\) b/b0. At 260 °C, the a, b, and c lattice parameters significantly decreased. Above 280 °C, hydromagnesite underwent a structural collapse with dehydration and dehydroxylation reactions, but was never accompanied by nucleation and growth of crystal phases up to 425 °C. During the thermal decomposition from hydromagnesite to periclase, the Mg atoms maintained the octahedral coordination environments in the structure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mechanisms of carbonate mineral formation and its phase change in geological system have been studied for many years because of their abundance and wide distribution over the Earth's crust. Magnesite (MgCO3), calcite (CaCO3), and dolomite [CaMg(CO3)2] are the stable phases at ambient conditions. Petrologic observations, laboratory experiments, and theoretical investigations on stability and breakdown kinetics of the carbonates have clarified that magnesite is the most stable carbonate phase throughout most of the mantle and can be subduct to the deep mantle due to its higher melting temperature [1,2,3,4,5,6,7]. Understanding mineralization processes of magnesite provides insight into the long-standing magnesite problem, which is defined by the difficulty to incorporate unhydrated magnesium ions into carbonate from aqueous solutions without changing the temperature or pressure [8,9,10,11,12,13,14,15,16]. Instead of precipitation of magnesite, a variety of metastable magnesium carbonate hydrates, pokrovskite [Mg2(CO3)(OH)2], nesquehonite [MgCO3·3H2O], lansfordite [MgCO3·5H2O], hydromagnesite [Mg5(CO3)4(OH)2·4H2O], dypingite [Mg5(CO3)4(OH)2·5H2O], and artinite [Mg2CO3(OH)2·3H2O], are mineralized in the MgO − CO2 − H2O system depending on the temperature, partial pressure of CO2, and pH [12, 17,18,19,20,21,22]. In an aqueous solution in the temperature range from room temperature to 55 °C and partial pressure of CO2 close to ambient pressure or below, nesquehonite, MgCO3·3H2O, is precipitated [23, 24]. Since nesquehonite is, however, unstable at near surface ambient conditions, it subsequently transforms to hydromagnesite, Mg5(CO3)4(OH)2⋅4H2O, at moderate temperature and CO2 pressure depending on the reaction time [23, 25, 26]. This is achieved through one or more dissolution–precipitation steps, giving rise to a variety of short-lived metastable intermediates such as dypingite-type phase, Mg5(CO3)4(OH)2·XH2O (X = 5, 6, 8, 11) [21, 26]. Hence, nesquehonite acts as a precursor for hydromagnesite, which is by far the most common naturally occurring magnesium carbonate hydrates minerals at atmospheric CO2 pressure within the temperature interval typical of most surface environment [21, 27]. With the further increase in the hydrothermal reaction temperature ranging from 120 to 185 °C, magnesite is formed via the dissolution–precipitation process [28, 29].
During the past decades, extensive research has been conducted on the thermal decomposition of hydromagnesite mainly by the TG/DTA technique [30,31,32,33,34,35,36,37]. From these studies, it has been clarified that the thermal decomposition of hydromagnesite is affected by atmospheric conditions, particularly the partial pressure of carbon dioxide. Below a partial pressure of carbon dioxide of 0.1 atm, only two decomposition steps are observed, the loss of the crystalline water, followed by the combined dehydroxylation and decarbonation [38, 39]. Under conditions of high partial pressure of carbon dioxide, on the other hand, the dehydroxylation and decarbonation occur separately, giving three steps [40]. During this decomposition process, the dehydrated hydromagnesite partially decarbonates to form magnesite [38, 39]. According to the sole report on the thermal decomposition of hydromagnesite by in situ high-temperature X-ray diffraction technique [41], crystal structure of hydromagnesite was essentially maintained up to 300 °C, but a small trace of magnesite also emerged at 300 °C. Diffraction peaks of the magnesite persisted up to at least 400 °C. With further increasing temperature to 500 °C, the diffraction peaks of both hydromagnesite and magnesite totally disappeared and those of periclase appeared. Ballirano et al. [42] have, however, insisted that hydromagnesite never transforms into magnesite because it possesses the Mg:CO3 molar ratio of 5:4, which is different from that of 1:1 in magnesite.
So far, there is still little information about crystal structure changes of hydromagnesite as well as its local structure changes during the thermal decomposition. Understanding of phase changes, crystal structure changes, and local structure changes on thermal decomposition process of hydromagnesite may provide insight into the long-term geological storage of carbon dioxide. In the study, hence, we investigated the thermal decomposition of hydromagnesite by in situ high-temperature XRD and ex situ high-temperature X-ray total scattering measurements. The structural changes during this thermal decomposition process were studied by using Rietveld refinement and atomic pair distribution function (PDF) analyses. The novelty of this study lies in the finding that hydromagnesite was anisotropically expanded with temperature and the local structure around Mg was little affected during the thermal decomposition. In the paper, we report temperature dependence of crystal structure of hydromagnesite and emerging phases produced by thermal decomposition of hydromagnesite in detail.
Experimental
Commercially available MgCl2 (Wako Pure Chemicals Co., Inc., purity ≥ 97.0%) and Na2CO3 (Wako Pure Chemicals Co., Inc., purity ≥ 99.5%) were used as starting materials. Hydromagnesite was hydrothermally synthesized using ultra-pure water (Milli-Q). First, 0.5 M Na2CO3 solution (25 mL) was added to 0.5 M MgCl2 solution (25 mL) and stirred at room temperature for 10 min. Second, the white slurry was filled in a Teflon vessel and sealed within a stainless-steel autoclave. It was then placed in an electric furnace held at 120 ± 1 °C for 1 day. After cooling down to room temperature with cold water, the resulting product was filtered through a filter paper. Finally, it was washed with Milli-Q water twice and then dried in air overnight. Hydromagnesite obtained in the study exhibited a typical spherical aggregate morphology with a particle size from 30 to 50 μm (Fig. 1). The result of X-ray diffraction measurement revealed that the product consisted entirely of hydromagnesite, together with a small amount of magnesite.

The SEM image of synthesized hydromagnesite
Before in situ high-temperature X-ray diffraction study, thermogravimetric (TG) and differential thermal analysis (DTA) were carried out using a TG/DTA-7300 thermal analyzer (Seiko Instruments Inc., Japan). Approximately 10 mg sample and α-alumina used as a reference were placed in Pt pans and heated over the temperature range from 60 to 600 °C with a heating rate of 10 °C min−1 in nitrogen flowing at 100 mL min−1.
In situ high-temperature X-ray diffraction patterns were obtained using an X-ray diffractometer (SmartLab, Rigaku Corp., Japan) equipped with a 9 kW X-ray generator (Cu Kα1 radiation λ = 1.54056 Å) and one-dimensional detector. High-temperature experiments were performed using a Pt heater used as a sample holder. The sample was mounted on a Pt sample holder and heated under a nitrogen atmosphere. The temperature was controlled with an accuracy of ± 1 °C by an R-thermocouple unit attached to the sample holder. The temperature was elevated to each target temperature at 10 °C min−1. After reaching the target temperature, it was maintained for 5 min before the measurements. The X-ray diffraction patterns were recorded in the 2θ range from 5° to 60° with a scanning speed of 2° min−1 in steps of 0.02°. Those for Rietveld refinement were collected in the 2θ range from 8° to 100°. The crystal structures of hydromagnesite were refined by the Rietveld method using the General Structure Analysis System (GSAS) package [43] with the EXPGUI interface [44]. Starting atomic coordinates for hydromagnesite were taken from the previous report [45]. Since hydrogen atoms were not included in the calculations, water molecules were refined as oxygen atoms in the refinements. The Rietveld refinement results are presented in Tables 1 and 2. Selected bond lengths and angles are summarized in Table 3. Representative Rietveld fitting profile is given in Fig. 2.

The result of Rietveld refinement profile
X-ray total scattering experiments for PDF analysis were conducted to investigate the local structure change during the thermal decomposition process from hydromagnesite to periclase through amorphous magnesium carbonate phase. High-energy X-ray diffraction measurements were conducted using a κ-type multi-axis diffractometer at BL14B1 bending magnet beamline in SPring-8, Japan. An incident X-ray beam was monochromatized using a Si(111) double-crystal monochromator to the wavelength of 0.205845(2) Å, because high Q (Q = 4πsinθ/λ) is critically important for the application of the PDF technique to obtain higher real-space resolution. Before the measurements, the samples were thermally treated in an electric furnace at the given temperatures (150 °C, 300 °C, 350 °C, and 425 °C) for an hour and then immediately quenched with liquid nitrogen. Samples of 50.00 mg were loaded in to Kapton capillary tubes with a 2 mm inner diameter and 15 mm length. A germanium point detector was scanned up to a wavenumber of Q = 25 Å−1. The PDFs were extracted from the one-dimensional diffraction patterns using the program PDFgetX2 [46]. The data were corrected for background scattering, sample absorption, X-ray polarization, Compton scattering, and normalization by average atomic scattering power to yield the total scattering structure function, S(Q). The Fourier transform of the S(Q) yielded the atomic pair distribution function, G(r). The obtained PDF profiles were analyzed using the program PDFgui [47].
Results and discussion
Thermal decomposition of hydromagnesite
Hydromagnesite crystalized in space group P21/c with lattice parameters a = 10.1217(5) Å, b = 8.9536(3) Å, c = 8.3886(2) Å, and β = 114.372(4) ° (Table 1). The Rietveld refinement converged to a satisfactory fit with the experimental data with the reliability indices Rp = 4.15%, wRp = 5.59%, and χ2 = 4.814. The bond lengths and angles in MgO6 octahedra and CO3 groups obtained in the study were essentially in agreement with those of the previous report [45]. Figure 3 shows crystal structure of hydromagnesite. The main structural motif was a zigzag ribbon of edge-sharing MgO6 octahedra at Mg1 and Mg11 sites running along the c-axis. The zigzag ribbons were connected together by corner-sharing MgO6 octahedra at Mg2 site to form a two-dimensional undulating sheet stacked through CO3 groups along the b-axis. In the structure, the MgO6 octahedra consisted of two distinct coordination environments. The MgO6 octahedra at Mg1 and Mg11 sites were surrounded by four oxygen atoms from CO3 groups, one hydroxyl group, and one water molecule. The MgO6 octahedron at Mg2 site was, on the other hand, surrounded by four oxygen atoms from CO3 groups and two hydroxyl groups. According to Ren et al. [37], the thermal decomposition process of hydromagnesite, Mg5(CO3)4(OH)2⋅4H2O, is considered as follows: As the initial stage of the decomposition reaction, the H2O molecules are removed from the MgO6 octahedra at Mg1 and Mg11 sites (loss of crystalline water). The resulting chemical composition in this stage corresponds to 4MgCO3·Mg(OH)2. As the temperature further increases, the hydroxyl groups bridging the zigzag ribbons start to be removed (releasing water), resulting in the chemical composition 4MgCO3⋅MgO. When the temperature reaches a certain value, the C–O bond begins to be disrupted and decarbonated (releasing carbon dioxide). Thus, this stage belongs to the nucleation and nuclei growth of periclase.

Crystal structure of hydromagnesite characterized by a zigzag ribbons of edge-sharing MgO6 octahedra along the c-axis, forming b two-dimensional undulating sheets stacked through CO3 groups along the b-axis
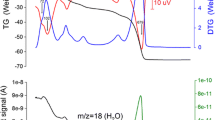
Figure 4 exhibits the TG/DTA curves for the thermal decomposition of hydromagnesite. The TG/DTG and the DTA curves exhibited two clear mass loss steps and two endothermic peaks at 263 °C and 445 °C, respectively. The TG/DTA curves observed in the study were in good agreement with the previous researches [35, 37, 38]. Table 4 exhibits the thermogravimetric date for hydromagnesite. The first mass loss of 16.4% was somewhat higher than the theoretical value of 15.4% corresponding to the elimination of 4 mol H2O from hydromagnesite. The second mass loss of 40.4% was, on the contrary, slightly lower than the theoretical mass loss of 41.5% corresponding to the elimination of 1 mol H2O and 4 mol CO2 from the dehydrated hydromagnesite. The result therefore suggested that dehydroxylation reaction starts when the dehydration reaction occurs.

The TG and DTA curves for the thermal decomposition of hydromagnesite
Phase change during the thermal decomposition
Variation of X-ray diffraction pattern of hydromagnesite with temperature is shown in Fig. 5. From the in situ X-ray diffraction data in the temperature range from 25 to 525 °C, it is possible to consider the evolution of hydromagnesite with temperature. Under a nitrogen atmosphere, no significant change in the diffraction pattern was observed up to 260 °C, although the dehydration of hydromagnesite must occur. A preliminary simulation result of powder X-ray diffraction pattern by Crystal Diffract (CrystalMaker Software Ltd.) suggested that the site occupancy factors of water oxygen atoms, i.e., OW1 and OW11, were closely related to the relative intensity for the 011 and 022 reflections, corresponding to the strongest and the second strongest peaks of hydromagnesite. In the present study, actually, these peaks observed at 30.8° and at 15.2° became weaker as temperature increases (Fig. 5). In particular, the intensities clearly decreased above 200 °C. In general, diffraction intensity must be affected by temperature (Debye–Waller) factor as well, but the dehydration reaction in hydromagnesite may be visually monitored from the decrease in relative intensity for the 011 and 022 reflections. Most of diffraction peaks of hydromagnesite became smaller at 260 °C and then disappeared at 280 °C. The resulting composition of amorphous magnesium carbonate inferred from the result of TG/DTA analysis was 4MgCO3⋅Mg(OH)2 or 4MgCO3⋅MgO. The losses of water molecules and hydroxyl ions were accompanied by a structural collapse of hydromagnesite. Li et al. [41] previously reported that a small trace of magnesite was formed during the thermal decomposition of hydromagnesite. In the present study, diffraction peaks corresponding to magnesite were observed as well (Fig. 5). Here, we must consider whether or not the magnesite observed up to 500 °C was formed by the thermal decomposition of hydromagnesite. The intensity of 104 reflection from magnesite was compared with that of 111 reflection from the Pt sample holder. As a result, the intensity ratio of magnesite 104 to Pt 111 remained essentially unchanged. Namely, there was no temperature dependence of the relative intensity between magnesite and Pt. We therefore finally concluded that no magnesite was formed through the thermal decomposition of hydromagnesite and that the magnetite observed in XRD pattern was originally mixed with hydromagnesite before the measurement. In the previous study by Li et al. [41], the magnesite seems to be present from the beginning of the experiment. Thus, their statement that magnesite is formed during the thermal decomposition process is highly suspect. In the present study, therefore, we concluded that hydromagnesite underwent a structural collapse with dehydration and dehydroxylation reactions, but was never accompanied by nucleation and growth of crystal phases. The amorphous magnesium carbonate was predominant over a wide temperature range from 280 to 400 °C. After the decarbonation reaction, new peaks corresponding to periclase (MgO) emerged and increased continuously up to 525 °C, but the X-ray diffraction peaks of periclase had a large full-width at half-maximum (FWHM) values, indicating a characteristic poor crystalline quality.

Variation of in situ high-temperature X-ray powder diffraction pattern in the temperature range from 25 to 525 °C. The Pt peaks are derived from the sample holder
Thermal expansion and crystal structural changes in hydromagnesite
Figure 6 shows the variations of lattice parameters of hydromagnesite as a function of temperature up to 260 °C. The result of the thermal analysis showed that hydromagnesite started losing mass gradually with increasing temperature (Fig. 4). In spite of the fact that the H2O molecules was released continuously with increasing temperature, the unit cell volume monotonously increased with temperature up to 220 °C. Linear thermal expansion coefficient of hydromagnesite between 25 and 220 °C calculated by the approximate equation, α = \(\frac{1}{V} \cdot \left( {\frac{\partial V}{{\partial T}}} \right)_{{\text{p}}}\), is 2.03 × 10–5 K−1. This thermal expansion coefficient was smaller than that of 3.92 × 10–5 K−1 for magnesite MgCO3 [48]. The linear thermal expansion coefficient of nesquehonite MgCO3⋅3H2O is, on the other hand, 9.2 × 10–5 K−1 [49]. This extremely large value is ascribed to the structural feature of nesquehonite consisting of infinite ribbons running along the b axis, which are interconnected via hydrogen bonds only [49]. Compared with the structural feature of nesquehonite, the zigzag ribbons in hydromagnesite are directly connected with MgO6 octahedra and CO3 groups (Fig. 3). Therefore, hydromagnesite never shows the large thermal expansion coefficient. The unit cell of hydromagnesite was anisotropically expanded as temperature increases. The c lattice parameter was monotonously increased with temperature up to 220 °C. The a lattice parameter also showed an increase trend with temperature. On the other hand, the b lattice parameter and β angle did not show a regular trend with temperature, suggesting that thermal expansion effect on the b lattice parameter and β angle was very small. The anisotropic thermal expansion is associated with the two-dimensional undulating sheets composed of edge-sharing MgO6 octahedra (Fig. 3).

The variation of unit cell of a, b, c, β, and V of hydromagnesite in the temperature range from 25 to 260 °C
Comparison of bond lengths and angles at temperatures between 25 and 220 °C is given in Table 3. Surprisingly, the mean bond lengths within MgO6 octahedra and CO3 groups at 220 °C were essentially identical to those observed at 25 °C within the standard deviations. The MgO6 octahedra and CO3 groups were anisotropically distorted from the regular octahedral and regular triangle configurations. In the hydromagnesite, the strongest hydrogen bond can be found between H4 and O11 at distances of 1.78(6) Å [45], which is oriented approximately along the [101] direction (Fig. 7). Since hydrogen bonds are weakened as temperature increases, the interatomic distance between H4 and O11 must become longer at high temperature. It must be therefore accompanied by an increase in the interatomic distance between the O11 and the OW11 directly bonded to the H4. In the study, the interatomic distance between the O11 the OW11 was certainly increased from 2.671(9) to 2.697(9) Å with temperature, which would lead to the expansion along the a- and c-axes. The relative expansion between 25 and 220 °C followed the c/c0 > a/a0 \(\gg\) b/b0. At 260 °C, however, the a, b, and c lattice parameters significantly decreased, resulting in the remarkable unit cell volume contraction. The fact that the unit cell volume at 260 °C was smaller than that observed at 25 °C supported the evidence that the effect of dehydration became relatively more pronounced than the thermal expansion effect.

The strongest hydrogen bond between H4 and O11, oriented approximately along the [101] direction
Structural modification in the amorphous magnesium carbonate
Figure 8 exhibits variation of the PDF patterns during the thermal decomposition of hydromagnesite. The PDF patterns indicate the local structure change during the thermal decomposition from hydromagnesite to periclase through the amorphous magnesium carbonate. The PDF pattern of hydromagnesite at room temperature exhibited interatomic correlation functions corresponding to the crystal structure of hydromagnesite. Since the C–O and Mg–O bond lengths in hydromagnesite ranged from 1.180(15) to 1.341(14) Å and from 1.992(11) to 2.160(10) Å, respectively (Table 3), the interatomic correlation peaks observed around 1.3 Å and 2.1 Å can be assigned to C–O bond and Mg–O bonds, respectively. Furthermore, the Mg–Mg distance between the edge-sharing MgO6 octahedra at Mg1 and Mg11 sites was 3.083(10) Å. The peaks at 3.0 Å can be therefore assigned to Mg–Mg distance. Based on the results of in situ high-temperature X-ray diffraction study and TG/DTA analysis, it can be concluded that by dehydration and dehydroxylation hydromagnesite was transformed to the amorphous magnesium carbonate between 280 and 400 °C. With decarbonation reaction, it was finally crystallized to periclase. The present study yielded several noteworthy findings. First, at 300 °C the interatomic correlation peak of C–O bond was obviously strengthened. The strong C–O peak at 300 °C would be derived from locally ordered CO3 groups in the amorphous magnesium carbonate. Second, the peak positions at 2.1 Å and 3.0 Å remained unchanged throughout the thermal decomposition. The Rietveld refinement result showed that the mean bond lengths within C–O and Mg–O bonds observed at 220 °C were identical to those observed at 25 °C, indicating that in hydromagnesite the coordination environments of Mg and C atoms were little affected with temperature. Moreover, the PDF analysis result also indicated that the octahedral coordinations around Mg atoms survived from hydromagnesite to periclase through the amorphous magnesium carbonate. Since intensity of interatomic correlation peaks must be related to the atomic pair density, the increase in the peak intensities can be explained by an increase in concentration of MgO6 octahedron in the structure.

The variation of PDF patterns during the thermal decomposition of hydromagnesite
Conclusions
The unit cell of hydromagnesite was anisotropically expanded with temperature up to 220 °C. The c lattice parameter was monotonously increased with temperature up to 220 °C. The a lattice parameter also showed an increase trend with temperature. The b lattice parameter and β angle, on the other hand, did not show a regular trend with temperature, suggesting that thermal expansion effect on the b lattice parameter and β angle was very small. The anisotropic thermal expansion was associated with the two-dimensional undulating sheets composed of edge-sharing MgO6 octahedra. The relative expansion between 25 and 220 °C followed the c/c0 > a/a0 \(\gg\) b/b0. At 260 °C, the a, b, and c lattice parameters significantly decreased. As a result, the unit cell volume became smaller than that observed at 25 °C, which suggested that the effect of dehydration became relatively more pronounced than the thermal expansion effect. The present study confirmed that under a nitrogen atmosphere hydromagnesite was decomposed to amorphous magnesite carbonate by dehydration and dehydroxylation reactions. The losses of water molecules and hydroxyl ions caused a structure collapse of hydromagnesite, but were never accompanied by nucleation and growth of crystal phases. The resulting amorphous magnesium carbonate was thermally stable over a wide temperature range from 280 to 400 °C. With decarbonation reaction, it was crystallized into periclase (MgO) above 425 °C. The X-ray diffraction peaks of periclase had large FWHM values, indicating a characteristic poor crystalline quality. During the thermal decomposition from hydromagnesite to periclase through the amorphous magnesite carbonate, the Mg atoms maintained the octahedral coordination environments in the structure.
References
Katsura T, Ito E. Melting and subsolidus phase relations in the MgSiO3–MgCO3 system at high pressures: implications to evolution of the Earth's atmosphere. Earth Planet Sci Lett. 1990;99:110–7.
Martinez I, Chamorro Peréz EM, Matas J, Gillet P, Vidal G. Experimental investigation of silicate–carbonate system at high pressure and high temperature. J Geophys Res. 1998;103:5143–63.
Isshiki M, Irifune T, Hirose K, Ono S, Ohishi Y, Watanuki T, Nishibori E, Takata M, Sakata M. Stability of magnesite and its high-pressure form in the lowermost mantle. Nature. 2004;427:60–3.
Sandengen K, Josang LO, Kaasa B. Simple method for synthesis of magnesite (MgCO3). Ind Eng Chem Res. 2008;47:1002–4.
Dasgupta R, Hirschmann MM. The deep carbon cycle and melting in Earth's interior. Earth Planet Sci Lett. 2010;298:1–13.
Rohrbach A, Schmidt MW. Redox freezing and melting in the Earth's deep mantle resulting from carbon–iron redox coupling. Nature. 2011;472:209–12.
Solopova NA, Dubrovinsky L, Spivak AV, Litvin YA, Dubrovinskaia N. Melting and decomposition of MgCO3 at pressures up to 84 GPa. Phys Chem Miner. 2014;42:73–81.
Sayles FL, Fyfe WS. The crystallization of magnesite from aqueous solution. Geochim Cosmochim Acta. 1973;37:87–99.
Königsberger E, Königsberger L, Gamsjager H. Lowtemperature thermodynamic model for the system Na2CO3-MgCO3-CaCO3-H2O. Geochim Cosmochim Acta. 1999;63:3105–19.
Pokrovsky OS, Schott J, Thomas F. Processes at the magnesium-bearing carbonates/solution interface. I. A surface speciation model for magnesite. Geochim Cosmochim Acta. 1999;63:863–80.
Xiong YL, Lord AS. Experimental investigations of the reaction path in the MgO–CO2–H2O system in solutions with various ionic strengths, and their applications to nuclear waste isolation. Appl Geochem. 2008;23:1634–59.
Hänchen M, Prigiobbe V, Baciocchi R, Mazzotti M. Precipitation in the Mg-carbonate system—effects of temperature and CO2 pressure. Chem Eng Sci. 2008;63:1012–28.
Sadly GD, Jordan G, Schott J, Oelkers EH. Magnesite growth rate as a function of temperature and saturation state. Geochim Cosmochim Acta. 2009;73:5646–57.
Bénézeth P, Saldi GD, Dandurand JL, Schott J. Experimental determination of the solubility product of magnesite at 50 to 200 °C. Chem Geol. 2011;286:21–31.
Chaka AM, Felmy AR. Ab initio thermodynamic model for magnesium carbonates and hydrates. J Phys Chem A. 2014;118:7469–88.
Qafoku O, Dixon DA, Rosso KM, Schaef HT, Bowden ME, Arey BW, Felmy AR. Dynamics of magnesite formation at low temperature and high pCO2 in aqueous solution. Environ Sci Technol. 2015;49:10736–44.
Perchiazzi N, Merlino S. The malachite-rosasite group: crystal structures of glaukosphaerite and pokrovskite. Eur J Mineral. 2006;18:787–92.
Back ME, Mandarino JA. Fleischer's glossary of mineral species. Tucson: Mineralogical Record Inc; 2008.
Frost RL, Bahfenne S, Graham J, Reddy BJ. The structure of selected magnesium carbonate minerals - A near infrared and mid-infrared spectroscopic study. Polyhedron. 2008;27:2069–76.
Beinlich A, Austrheim H. In situ sequestration of atmospheric CO2 at low temperature and surface cracking of serpentinized peridotite in mine shafts. Chem Geol. 2012;332:32–44.
Hopkinson L, Kristova P, Rutt K, Cressey G. Phase transitions in the system MgO–CO2–H2O during CO2 degassing of Mg-bearing solutions. Geochim Cosmochim Acta. 2012;76:1–13.
Kristova P, Hopkinson LJ, Rutt KJ, Hunter HMA, Cressey G. Carbonate mineral paragenesis and reaction kinetics in the system MgO-CaO-CO2-H2O in presence of chloride or nitrate ions at near surface ambient temperatures. Appl Geochem. 2014;50:16–24.
Zhang Z, Zheng Y, Ni Y, Liu Z, Chen J, Liang X. Temperature- and pH-dependent morphology and FT−IR analysis of magnesium carbonate hydrates. J Phys Chem B. 2006;110:12969–73.
Morgan B, Wilson SA, Madsen IC, Gozukara YM, Habsuda J. Increased thermal stability of nesquehonite (MgCO3·3H2O) in the presence of humidity and CO2: implications for low-temperature CO2 storage. Int J Greenhouse Gas Control. 2015;39:366–76.
Davies PJ, Bubela B. The transformation of nesquehonite into hydromagnesite. Chem Geol. 1973;12:289–300.
Hopkinson L, Rutt K, Cressey G. The transformation of nesquehonite to hydromagnesite transition in the system MgO–CaO–H2O–CO2: an experimental spectroscopic study. J Geol. 2008;116:387–400.
Langmuir D. Stability of carbonates in the system MgO−CO2–H2O. J Geol. 1965;73:730–54.
Di Lorenzo F, Rodriguez-Galan RM, Prieto M. Kinetics of the solvent-mediated transformation of hydromagnesite into magnesite at different temperatures. Mineral Mag. 2014;78:1363–72.
Farhang F, Oliver TK, Rayson M, Bren G, Stockenhuber M, Kennedy E. Experimental study on the precipitation of magnesite from thermally activated serpentine for CO2 sequestration. Chem Eng J. 2016;303:439–49.
Botha A, Strydom CA. Preparation of a magnesium hydroxy carbonate from magnesium hydroxide. Hydrometallurgy. 2001;62:175–83.
Haurie L, Fernandez AI, Velasco JI, Chimenos JM, Cuesta JML, Espiell F. Synthetic hydromagnesite as flame retardant Evaluation of the flame behaviour in a polyethylene matrix. Polym Degrad Stab. 2006;91:989–94.
Vagvolgyi V, Frost RL, Hales M, Locke A, Kristof J, Horvath E. Controlled rate thermal analysis of hydromagnesite. J Therm Anal Calorim. 2008;92:893–7.
Hollingbery LA, Hull TR. The thermal decomposition of huntite and hydromagnesite—a review. Thermochim Acta. 2010;509:1–11.
Hollingbery LA, Hull TR. The thermal decomposition of natural mixtures of huntite and hydromagnesite. Thermochim Acta. 2012;528:45–52.
Bhattacharjya D, Selvamani T, Mukhopadhyay I. Thermal decomposition of hydromagnesite effect of morphology on the kinetic parameters. J Therm Anal Calorim. 2012;107:439–45.
Unluer C, Al-Tabbaa A. Characterization of light and heavy hydrated magnesium carbonates using thermal analysis. J Therm Anal Calorim. 2014;115:595–607.
Ren HR, Chen Z, Wu YL, Yang MD, Chen J, Hu HS, Liu J. Thermal characterization and kinetic analysis of nesquehonite, hydromagnesite, and brucite, using TG-DTG and DSC techniques. J Therm Anal Calorim. 2014;115:1949–60.
Sawada Y, Uematsu K, Mizutani N, Kato M. Thermal decomposition of hydromagnesite 4MgCO3-Mg(OH)2–4H2O under different partial pressures of carbon dioxide. Thermochim Acta. 1978;27:45–59.
Sawada Y, Yamaguchi J, Sakurai O, Uematsu K, Mizutani N, Kato M. Thermal decomposition of basic magnesium carbonates under high-pressure gas atmospheres. Thermochim Acta. 1979;32:277–91.
Padeste C, Oswald HR, Reller A. The thermal behavior of pure and nickel-doped hydromagnesite in different atmospheres. Mater Res Bull. 1991;26:1263–8.
Li Q, Ding Y, Yu GH, Li C, Li FQ, Qian YT. Fabrication of light-emitting porous hydromagnesite with rosette-like architecture. Solid State Commun. 2003;125:117–20.
Ballirano P, De Vito C, Mignardi S, Ferrini V. Phase transitions in the Mg-CO2-H2O system and the thermal decomposition of dypingite, Mg5(CO3)4(OH)2·5H2O: implications for geosequestration of carbon dioxide. Chem Geol. 2013;340:59–67.
Larson AC, Von Dreele RB. General Structure Analysis System, (GSAS). Los Alamos National Laboratory Report LAUR 86–784; 2004.
Toby BH. EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr. 2004;34:210–3.
Akao M, Iwai S. The hydrogen bonding of hydromagnesite. Acta Crystallogr B. 1977;33:1273–5.
Qui X, Thompson JW, Billinge SJL. PDFgetX2: A GUI driven program to obtain the pair distribution function from X-ray powder diffraction data. J Appl Crystallogr. 2004;37:678.
Farrow CL, Juhas P, Liu JW, Bryndin D, Božin ES, Bloch J, Proffen T, Billinge SJL. PDFfit2 and PDFgui: computer programs for studying nanostructure in crystals. J Phys Condens Matter. 2007;19:335219.
Wang ML, Shi GH, Qin JQ, Bai Q. Thermal behaviour of calcite-structure carbonates: a powder X-ray diffraction study between 83 and 618K. Eur J Mineral. 2018;30:939–49.
Ballirano P, De Vito C, Ferrini V, Mignardi S. The thermal behaviour and structural stability of nesquehonite, MgCO3⋅3H2O, evaluated by in situ laboratory parallel-beam X-ray powder diffraction: New constraints on CO2 sequestration within minerals. J Hazard Mater. 2010;178:522–8.
Acknowledgements
This work was performed under the Shared Use Program of JAEA Facilities (Proposal No. 2018A-E06) with the approval of Nanotechnology Platform project supported by the Ministry of Education, Culture, Sports, Science and Technology (Proposal No. A-18-AE-0005). The synchrotron radiation experiments were performed at JAEA beamline BL14B1 in SPring-8 (Proposal No. 2018A3633). The work was partially supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (Project No. 17K05702).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yamamoto, Gi., Kyono, A., Sano, Y. et al. In situ and ex situ studies on thermal decomposition process of hydromagnesite Mg5(CO3)4(OH)2·4H2O. J Therm Anal Calorim 144, 599–609 (2021). https://doi.org/10.1007/s10973-020-09618-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09618-7