
Abstract
Spinel ferrites are the technologically important materials whose application ranges from the environmental, bio-medical to the smart devices. The properties of the ferrite can be tuned by varying the particle size of the different metal ion substituted in the core of the ferrite. In the present studies, we have successfully prepared sparingly investigated Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrite series by precursor combustion method using nickel–manganese–zinc–iron fumarato–hydrazinate as precursor. These metal fumarato–hydrazinate precursors burn auto-catalytically, once ignited with a burning splinter, to form metal ferrites. The detailed study of these precursors was carried out using TG–DTA, IR, CHN analysis, isothermal mass loss and total mass loss studies. The experimental hydrazine content as well as contents of C, H, N, Ni, Mn, Zn and Fe of the precursors matched exactly with the theoretical values. The thermal analysis of the precursors suggest mainly two major steps decomposition. The XRD patterns and the IR spectra of the ferrites obtained by burning these precursors confirmed the single-phase cubic nature of these ferrites. The resistivity studies indicate decrease in resistivity with increase in Mn2+ content of the ferrites. The Curie temperature also shows the same trend.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Spinel ferrites are one of the most widely studied compounds because of their varied applications in different fields. One of their applications is, as transformer cores, which is because of their properties like high saturation magnetization, high resistivity and low core losses which in turn depends upon the method of preparation and the resultant size of the ferrites obtained [1]. With the advancement in preparation techniques, newer methods were developed in order to synthesize the ferrites in nano-size range. The nanosized transition metal oxides play a crucial role in almost all the fields. For example, it has been observed from the literature survey that these oxides play an important role as gas sensor, in air pollution control, in catalysis, in bio-medical devices as well as in the development of rechargeable accumulators [2,3,4,5,6]. They also find applications in targeted drug delivery, magnetic data storage, as a magneto-optic material, in energy conversion, in high performance digital tapes, refrigerators, ferrofluids [7,8,9], adsorption of dyes [10], etc.
Ni–Zn ferrite are the soft magnetic materials much like Co–Zn ferrites, with high resistivity and low permeability at higher frequency, whereas Mn–Zn ferrite are the materials possessing highest permeability and saturation induction making this combination favorable to work for high frequency magnetic applications [11,12,13,14]. Many researchers have reported the preparation of Ni–Mn–Zn ferrite using various methods. From the literature search, we found out that the research for the synthesis of Ni–Zn–Mn ferrite has started way back in 1983 wherein Goto et al. [15] employed Bridgman method to grow a single crystal of Mn-substituted Ni–Zn ferrite. A uniaxial hot pressing method using double action hydraulic press was used by Babbar et al. [16] to prepare Mn0.6Zn0.4−xNixFe2O4 (x = 0.00–0.4 in steps of 0.05). Ceramic method was the most favored method in the ancient days to synthesize the ferrites, and many researchers used this method to synthesize Ni–Mn–Zn ferrites. Bara et al. [17] have synthesized MnxZnyNizFe2O4 (x, y, z ranging from 0 to 1 in steps of 0.1) using this ceramic sintering method. Other researchers, such as Zhong et al. [18], have synthesized Ni0.44Zn0.56MnxFe2−xO4 (x = 0.00–0.15), Sattar et al. [19, 20] have synthesized Ni0.6−tMntZn0.4Fe2O4 (x = 0.0–0.6), Rao et al. [21] have synthesized Ni0.35Zn0.65−xMnxFe2O4 (x = 0.00–0.25), and Hossain et al. [22] have synthesized Ni0.50−xMnxZn0.50Fe2O4 (x = 0.00, 0.10 and 0.20) using this ceramic method. With time and the advancement in the facilities, the researchers moved on from the conventional solid state method to the non-conventional wet chemical methods to synthesize the ferrites. Singh et al. have used citrate precursor method to synthesize various Ni–Mn–Zn ferrite such as, MnxNi0.5−xZn0.5Fe2O4, (x = 0.05, 0.1, 0.2, 0.3, 0.4) [23,24,25,26], NixMn0.4−xZn0.6Fe2O4 (x = 0.1–0.4) [27], Ni0.3MnxZn0.7−xFe2O4 (x = 0.1–0.5) [28], Mn0.2ZnxNi0.8−xFe2O4 (x = 0.2–0.6) [29] and Mn0.2Ni0.2Zn0.6Fe2O4 [30]. The citrate precursor method has also been employed by other researchers to synthesize Ni–Mn–Zn ferrite [31,32,33,34,35]. The Sol–gel auto combustion method has also been adopted for the synthesis of Ni–Mn–Zn ferrite [1, 36,37,38,39,40,41]. Venkataraju et al. [42] have prepared Mn0.5−xNixZn0.5Fe2O4 (x = 0.0–0.3) using co-precipitation method, while Gawas et al. have employed fumarato–hydrazinate precursor method to synthesize Mn0.3Ni0.3Zn0.4Fe2O4 [43] and Ni0.5−xMnxZn0.5Fe2O4 (x = 0.0–0.5) [11, 44].
From the literature survey, it is revealed that the ferrite with composition Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) has been rarely studied [37], so we planned to synthesize this Ni0.7−xMnxZn0.3Fe2O4 series using the precursor combustion method which was first devised by our group [43,44,45,46,47,48,49]. In this work, we have employed fumarato–hydrazinate as a ligand and have synthesized Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6) precursors. Since the lifespan of these precursors is short, their characterization was done immediately once the precursor was prepared. The auto-catalytic thermal decomposition of these precursors gave Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites which were also characterized, and the results of the same are presented here along with precursors characterization results.
Experimental
Synthesis
Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites were prepared using the precursor combustion method. The detail preparation procedure has been described elsewhere [49]. In the present case, a solution of mixed metal chlorides was added to the solution containing requisite amount of sodium fumarate and hydrazine hydrate which was stirred for 2 h in N2 atmosphere (The amount of mixed metal chlorides taken is given in Table 1 of supplementary information). Upon addition of solution of mixed metal chlorides, a yellow colored precipitate was instantaneously formed, which was filtered, washed with ethanol and dried with diethyl ether. The characterization of these precursors was immediately carried out as they readily absorb moisture. After the characterization is done, a small part of these precursors was burned using a burning splinter. The precursors catch fire, which then spreads over the entire precursor, thereby auto-catalytically decomposing it to form ferrites in around 5 min. These Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites were then heated in furnace at 773 K for 5 h to remove any unburned carbonaceous species, if remained during auto-catalytic decomposition. These ferrites thus formed are called as the ‘as prepared’ ferrites. The ‘as prepared’ ferrites were characterized using XRD, IR and SEM/EDS techniques, and their electrical as well as magnetic properties were investigated.
Characterization techniques
The carbon, nitrogen and hydrogen content of the precursors were determined by using Elementar Vario Micro Cube CHNS analyzer. The TG–DTG–DTA analysis of the precursors was carried out using NETZSCH STA 409 PC (Luxx) analyzer in air atmosphere at a heating rate of 10 K per minute from RT-1073 K. The IR spectra of these precursors and the ferrites obtained from these precursors were recorded on the Shimadzu FTIR-IR prestigue-21 spectrophotometer from 350 to 4000 cm−1. The isothermal mass loss studies of these precursors were carried out at different set temperatures followed by their hydrazine estimation volumetrically under Andrew’s condition [50] using the standard 0.025 M KIO3 solution as titrant. The total mass loss studies of the precursor were carried out at 773 K for 5 h. The chemical analysis of all the ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites was carried out in order to find out the percentages of the metal ions present in them. The chemical analysis begins with the decomposition of these ferrites using 9 N HCl and then the separation of the metal ions on an ion-exchange column as described by Wilkins [51] and Kraus et al. [52]. The estimation of Ni2+, Mn2+ and Zn2+ was done complexometrically, while Fe3+ was estimated gravimetrically [50]. The X-ray diffraction studies of the ‘as prepared’ ferrites were carried out using Rigaku Miniflex X-ray diffractometer with Cu Kα radiations filtered through Ni absorber. The SEM/EDS studies were carried out on Carl Zeiss SEM model No. EVO18. The electrical resistivity of all the ‘as prepared’ ferrites was measured using two probe resistivity setup from RT to 823 K. The normalized AC susceptibility studies were carried out on AC susceptibility setup supplied by ADEC Embedded Technology and solutions Pvt. Ltd., Corlim, Goa-India from RT to 862 K.
Result and discussion
Characterization of precursors
Chemical analysis
The CHN analysis of all the precursors was carried out soon after their preparation. The C, H, N percentages in all the precursors obtained experimentally matches very well with the theoretically calculated percentages as can be seen from Table 1. The amount of C, H and N determined in all the precursors helps in fixing their formulae as Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6).
IR Studies
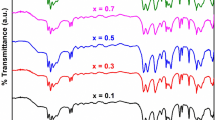
The infrared spectra of all the precursors are shown in Fig. 1a, b and we can observe that they are nearly identical. The absorption bands in the region 3067–3390 cm−1 in all the precursors correspond to the N–H stretching frequencies. The N–N stretching frequency observed at around 972 cm−1 indicates the bidentate bridging nature of the hydrazine ligand [48]. The absorption band observed in all the precursors at around 1626 cm−1 is due to C=C functional group in fumarate ion. The asymmetric and symmetric stretching frequencies of carboxylate ion in all the precursors were observed at 1549–1585 and 1370–1380 cm−1, respectively, with ∆ν(νas − νs) separation of 169–213 cm−1. This wide separation indicates the monodentate linkage of both carboxylate groups in the dianion [47]. Thus, IR data confirm the formation of all the precursors.

a, b IR spectra of Ni0.7−xZn0.3MnxFe2(C4H2O4)3·6N2H4 (x = 0.1–0.6)
TG–DTG–DTA analysis
The TG–DTG–DTA analysis of the precursors was carried out in order to study the decomposition pattern of the precursors. Figure 2a–f shows the TG–DTG–DTA curves of the Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6) precursors, while Table 2 shows the thermal decomposition data pertaining to these TG–DTG–DTA curves. In each of the cases, it can be seen that the precursors decompose into ferrites in 4 steps. Out of which two are major steps. The first minor step includes the beginning of dehydrazination with loss of either 0.5–1.5 hydrazine molecule followed by complete dehydrazination and starting of decarboxylation which is a major second step. The third step is also a major step which includes total decarboxylation, while the fourth minor step includes the formation of the ferrite with loss of any unburned species, if any.

a–f The TG–DTG–DTA curves of Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6)
The TG–DTG–DTA plot of Ni0.6Mn0.1Zn0.3Fe2(C4H2O4)3·6N2H4 (Fig. 2a) shows a mass loss of 5.33% from RT-380 K in TG corresponding to the loss of just little more than 1 hydrazine molecule followed by a major mass loss of 38.92% from 380 to 523 K corresponding to the loss of remaining little less than 5 hydrazine molecules indicating total dehydrazination followed by beginning of decarboxylation. The peak due to this mass loss has been observed in DTG at 388 K. An exothermic peak due to these reactions is observed in DTA plot at 405 K. The total decarboxylation takes place from 523 to 723 K with a mass loss of 22.12%. A broad exothermic peak due to decarboxylation is observed in DTA from 500 to 720 K, while in DTG, it is observed from 520 to 607 K. A small mass loss of around 2.56% due to the loss of unburned carbonaceous species was observed from 723 to 1073 K. The ferrite is formed at around 723 K with the residual mass of 33.63% in TG which matches closely with the theoretical mass loss of 33.40%.
Figure 2b shows the TG-DTG-DTA trace of Ni0.5Mn0.2Zn0.3Fe2(C4H2O4)3·6N2H4 precursor wherein the first mass loss of 4.32% indicating the loss of 1 hydrazine molecule is observed in TG from RT-358 K. From 358 to 523 K, a mass loss of 39.86% with an exothermic peak seen in DTA at 402 K and a peak in DTG at 365 K corresponds to the total dehydrazination followed by starting of decarboxylation. From 523 to 723 K, complete decarboxylation takes place which can be observed from mass loss of 20.44% in TG. An exothermic peak, in this temperature range, was observed at 589 K in both DTA and DTG trace. From 723 to 1073 K, a mass loss of 2.16% is observed which may be due to the loss of unburned organic moieties left. A total mass loss of 66.78% is observed in TG which matches closely with the theoretical mass loss of 66.63%.
In case of TG–DTG–DTA curve of Ni0.4Mn0.3Zn0.3Fe2(C4H2O4)3·6N2H4 (Fig. 2c), mass loss of 3.86% begins from RT up to 373 K which corresponds to loss due to just above half hydrazine molecule. This is followed by mass loss of 27.14% due to total dehydrazination and onset of decarboxylation in the temperature range of 373–523 K with an exothermic peak in DTA at 441 K and a peak in DTG at 425 K. A complete decarboxylation takes place from 523 to 673 K which can be evidenced from the mass loss of 34.61% and a broad exothermic peak in the region 520–690 K in DTA and at 563 K in case of DTG. A mass loss of 1.67% is observed from 673 to 1073 K due to the gradual loss of any unburned species remaining. The theoretical total mass loss for this precursor is 66.67% and is in close agreement with the observed mass loss of 67.28%.
In Fig. 2d (TG–DTG–DTA plot of Ni0.3Mn0.4Zn0.3Fe2(C4H2O4)3·6N2H4), it can be seen that a mass loss of 5.06% is observed from RT-376 K which matches up with the mass loss due to just little more than 1 hydrazine molecule. The remaining hydrazine molecules are lost in the subsequent step along with the beginning of decarboxylation with a mass loss of 42.75% in TG in the temperature range of 376–539 K. An exothermic peak at 417 K in DTA and a peak at 367 K in DTG is observed in the same region. From 539 to 723 K, a mass loss of 16.8% in TG and an exothermic peak in DTA at 583 K and a peak in DTG at 589 K, respectively, indicates the complete decarboxylation. A loss of unburned species with mass loss of 1.93% is observed from 723 to 1073 K. The total residue remained at 1073 K is 33.46% which matches closely with the theoretical value of 33.3%.
It can be observed from the TG–DTG–DTA curve of Ni0.2Mn0.5Zn0.3Fe2(C4H2O4)3·6N2H4 (Fig. 2e) that approximately 1 hydrazine molecule is lost from RT-379 K (a mass loss of 3.86% is observed in this temperature range) followed by a mass loss due to total dehydrazination and emergence of decarboxylation which can be inferred from the mass loss of 25.7% in the temperature range of 379–513 K with an exothermic peak at 437 K in DTA and a peak at 426 K in DTG. A mass loss of 35.06% from 513 to 673 K with an exothermic peak in DTA at 592 K and a peak at 574 K in DTG tallies with the mass loss due to total decarboxylation. From 673 to 1073 K, a small mass loss of 0.95% is observed which may be due to loss of any unburned species remaining. The total mass loss of 65.57% is nearly in close agreement with the theoretical mass loss of 66.74%.
Figure 2f depicts the TG–DTG–DTA curve of Ni0.1Mn0.6Zn0.3Fe2(C4H2O4)3·6N2H4 precursor. It can be noticed from this curve that from RT-389 K, there is a mass loss of 6.01% due to loss of nearly one and half hydrazine molecule. A small exo hump is observed in DTA at 368 K due to this loss. From 389 to 507 K, a mass loss of 24.03% is observed which corresponds to total dehydrazination and onset of decarboxylation. In DTA curve, an exothermic peak due to these reactions is observed at 441 K, while in DTG curve a peak is observed at 432 K. Total decarboxylation takes place from 507 to 673 K, which can be evidenced from the mass loss of 34.83% in TG and an exothermic peak at 589 K in DTA. DTG, however, shows a main peak at 581 K with a shoulder peak attached to main peak at 591 K indicating two-step decarboxylation. A mass loss of 1.18% from 673 to 1073 K is observed, which can be due to decomposition of unburned carbonaceous species. A total residue of 33.95% is obtained at the end which matches very well with the theoretical residue of 33.23%.
Thus, from the above thermal data, the correctness of the predicted formula for all the precursors is confirmed.
Isothermal mass loss studies, total mass loss and determination of hydrazine content
To compliment TG–DTG–DTA studies, the isothermal mass loss studies of the precursors were carried out at different set temperatures followed by the estimation of hydrazine. The data obtained from these isothermal mass loss and hydrazine estimation studies at different temperatures are presented in Table 3. The mass loss of 4.89%, 4.58%, 4.86% and 4.72% observed in precursors with composition x = 0.1, 0.2, 0.4 and 0.5, respectively, in the temperature region RT to 343 K corresponds to loss of moisture along with ~ 2–3% hydrazine. The composition x = 0.3 and 0.6, however, shows abnormally high (9.2%) and low (2.49%) mass loss values, respectively, may be due to excess of moisture and negligible amount of moisture in the precursors, respectively. The precursors in the temperature range of 343–373 K show mass loss from 4.04 to 5.82% due to loss of corresponding percentage of hydrazine in the precursors except x = 0.6 which shows exceptionally lower mass loss of 1.52% which matches with the hydrazine loss of 1.6%. However, in the temperatures from 373 to 408 K, composition x = 0.6 shows 12.66% loss in hydrazine percentage, while others show 7.04–11.1% hydrazine loss. All the precursors finally catch fire between 408 and 423 K.
The total mass loss studies were carried out by keeping the known amount of the precursor in muffle furnace at 773 K for 5 h. The total mass loss obtained experimentally matches well with the theoretical value (Table 3) thus further proving the formation of the precursor with the desired stoichiometry. For estimation of hydrazine content, as soon as the precursor was synthesized, a required amount of the precursor was weighed and added to 60% HCl and kept aside to determine the hydrazine content of the precursor. The estimation of the hydrazine was done by titrating against 0.025 M KIO3 solution [50]. The determined content of the hydrazine was found to be closely matching with the theoretical value (Table 3) which further strengthens the predicted formula for the precursor.
Characterization of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6)
Chemical analysis
The chemical analysis of all the ‘as prepared’ ferrites was carried out as described above in characterization techniques. The experimental and the theoretical amount of the metal ions present in Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) are tabulated in Table 4. It can be seen from this table that the theoretical and the experimental value in all the cases matches very well with each other confirming the formation of the ferrites with the desired stoichiometry.
Powder X-ray diffraction studies
The XRD patterns of all the ‘as prepared’ ferrite samples were recorded, and they are presented in Fig. 3. It can be seen from these XRD patterns that all the ‘as prepared’ ferrites show single spinel cubic phase without any impurities. The hkl planes corresponding to the cubic structure of spinel ferrites at (220), (311), (222), (400), (422), (333), (440) and (622) are observed in all the ferrite samples. Using this XRD patterns, the lattice parameter and the crystallite size of all the ‘as prepared’ ferrite samples were obtained and the same is given in Table 5. The lattice parameter of all the ferrite samples were obtained using the following equation
where ‘\(a\)’ is the lattice parameter and ‘d’ is the interplanar spacing given by the Bragg’s law for the (hkl) planes.

The XRD patterns of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6)
The crystallite size (t) of all the ‘as prepared’ ferrites was obtained using the Scherrer’s formula
where \(\lambda\) is the X-ray wavelength, \(\beta\) is the full width at half maxima and \(\theta\) is the Bragg’s diffraction angle.
From Table 5, it can be clearly observed that the lattice parameter goes on increasing with increase in Mn2+ concentration. This is because of the replacement of the smaller ion Ni2+(0.69 Å) with bigger ion Mn2+ (0.82 Å).
The XRD patterns indicate the nano-size nature of the crystallite particles. This observation is also supported by SEM studies. The crystallite size obtained varies in the range of 18–30 nm, with decrease in the crystallite size as Mn2+ concentration increases except for the sample with composition x = 0.4.
IR studies
The IR spectra of these ferrites are shown in Fig. 4. From Fig. 4, it can be concluded that there is absence of any organic moiety in these oxides. Only two peaks in IR spectra, typical of the spinel ferrites at around 400 and 600 cm−1, corresponding to the intrinsic stretching vibrations at octahedral and tetrahedral sites, respectively, confirm the formation of the spinel phase ferrites.

The IR spectra of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6)
SEM/EDS studies
The SEM images of these ferrites shown in Fig. 5 confirm the spherical nature of these particles which are agglomerated. From the histogram of the particle size distribution, it can be seen that the average particle size varies from 18 to 30 nm, complementing the results obtained using the Scherrer’s formula. The EDS mapping of ferrites confirms the presence of the required metal ions in the desired ratios in all the ferrite samples. The data pertaining to this are given in Table 4. It can be observed from this table that the theoretical and the experimental percentages of metal ions in the ferrites matches very well with each other. Only in case of iron, the EDS data show higher percentages which may be because of lower observed percentages of oxygen (Table 4). However, percentage of iron obtained by chemical analysis matches closely with theoretical percentages confirming the formation of stoichiometric ferrites as per the formulae.


The SEM images and EDS mapping of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6)
Electrical resistivity studies
The electrical resistivity studies of all the Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites were measured during both heating and cooling, and the plot of the log resistivity as a function of 1000/T is given in Fig. 6a, b. From Fig. 6a, b, we can observe that resistivity first slightly increases and then starts decreasing with increase in temperature during heating while during cooling cycle there is a gradual decrease in resistivity with increase in temperature indicating the normal behavior of semiconductors. The increase at lower temperature in resistivity during heating might be due to the presence of some impurities like moisture which when heated disappears, further leading in decrease in resistivity with increasing temperature. Also, from Fig. 6 we can observe that as the Mn2+ concentration is increasing the resistivity of the ferrite samples goes on decreasing. As we know from the literature that Ni–Zn ferrite has the higher resistivity, while Mn–Zn ferrites shows lower resistivity, so incorporation of Mn2+ in place of Ni2+ leads to decrease in resistivity of the ferrite samples. The same type of trend has been reported by Singh et al. [24].

The plot of log resistivity v/s 1/T * 1000 of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) during a heating and b cooling
AC susceptibility studies
The AC susceptibility studies of all the ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites were carried out in order to know the Curie temperature as well as the nature of the particles. The particles in the ferrite are normally present in three types of domains, namely, multi-domain, single domain and superparamagnetic. Based on the size of the particles, they exist in either single type or mixture of two types of particles. Based on the nature of the plot of normalized AC susceptibility versus temperature, the particles domain can be determined.
Figure 7 shows the plot of normalized AC susceptibility versus temperature of all the ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites. The temperature at which ferromagnetic sample converts to paramagnetic is called as the Curie temperature, and the same for all the ‘as prepared’ ferrites has been found out from this studies and is presented in Table 5. From Fig. 7, it can be observed that the Curie temperature of the ferrite samples goes on decreasing with increase in Mn2+ concentration and from the nature of the plots it can be concluded that all the ‘as prepared’ ferrites shows single-domain type of particles with increasing superparamagnetic character with increase in Mn2+ concentration. The relatively smaller crystallite size of Ni0.7−xMnxZn0.3Fe2O4 for composition x = 0.3, x = 0.5 and 0.6 also leads to increase in superparamagnetic type of domains which is clearly seen in Fig. 7.

The plot of normalized AC susceptibility as a function of temperature of ‘as prepared’ Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6)
Conclusion
Single-phase, nanosized Ni0.7−xMnxZn0.3Fe2O4 (x = 0.1–0.6) ferrites were synthesized using a low-cost precursor combustion method. The precursors synthesized during this method (Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6)) were thoroughly characterized using various instrumental and experimental techniques which helped in confirming the formula of the precursor. The novelty of these precursors is that they burn auto-catalytically to form ferrites, once initially ignited. Two major mass loss regions have been found in TG–DTA curves confirming two-step decomposition of these precursors to ferrites. The substitution of Mn2+ in Ni–Zn ferrites results in increase in lattice parameter but decrease in the resistivity and Curie temperature. From the nature of normalized AC susceptibility plots, it was observed that all the ferrites have single-domain particles with increase in the superparamagnetic character with increase in Mn2+ concentration in Ni–Zn Ferrites.
References
Ramesh S, Dhanalakshmi B, Sekhar BC, Rao PSVS, Rao BP. Effect of Mn/Co substitutions on the resistivity and dielectric properties of nickel-zinc ferrites. Ceram Int. 2016;42:9591–8.
Ştefãnescu M, Dippong T, Stoia M, Ştefãnescu O. Study on the obtaining of cobalt oxides by thermal decomposition of some complex combinations, undispersed and dispersed in SiO2 matrix. J Therm Anal Calorim. 2008;94:389–93.
Mukherjee K, Bharti DC, Majumder SB. Solution synthesis and kinetic analysis of the gas sensing characteristics of magnesium ferrite particles. Sens Actuators B. 2010;146:91–7.
Mukherjee K, Majumder SB. Synthesis process induced improvement on the gas sensing characteristics of nano-crystalline magnesium zinc ferrite particles. Sens Actuators B. 2012;162:229–36.
Sutka A, Mezinskis G, Lusis A, Stingaciu M. Gas sensing properties of Zn-doped p-type Nickel ferrite. Sens Actuators B. 2012;171-172:354–60.
Dippong T, Levei EA, Tanaselia C, Gabor M, Nasui M, Tudoran LB, Borodi G. Magnetic properties evolution of the CoxFe3-xO4/SiO2 system due to advance thermal treatment at 700 °C and 1000 °C. J Magn Magn Mater. 2016;410:47–54.
Dippong T, Levei EA, Cadar O. Preparation of CoFe2O4/SiO2 nanocomposites at low temperatures using short chain diols. J Chem. 2017. https://doi.org/10.1155/2017/7943164.
Stefanescu M, Stoia M, Caizer C, Dippong T, Barvinschi P. Preparation of CoxFe3-xO4 nanoparticles by thermal decomposition of some organo-metallic precursors. J Therm Anal Calorim. 2009;97:245–50.
Sharifi I, Shokrollahi H, Amiri S. Ferrite-based magnetic nanofluids used in hyperthermia applications. J Magn Magn Mater. 2012;324:903–15.
Liu R, Fu H, Yin H, Wang P, Lu L, Tao Y. A facile sol combustion and calcination process for the preparation of magnetic Ni0.5Zn0.5Fe2O4 nanopowders and their adsorption behaviors of Congo red. Powder Technol. 2015;274:418–25.
Gawas UB, Verenkar VMS, Meena SS, Bhatt P. Influence of Mn substitution on Mössbauer and magnetic properties of Ni-Zn ferrite nanoparticles. J Supercond Nov Magn. 2017;30:3241–6.
Dippong T, Levei EA, Cadar O, Deac IG, Diamandescu L, Barbu-Tudoran L. Effect of nickel content on structural, morphological and magnetic properties of NixCo1-xFe2O4/SiO2 nanocomposites. J Alloys Compd. 2019;786:330–40.
Dippong T, Deac IG, Cadar O, Levei EA, Diamandescu L, Borodi G. Effect of Zn content on structural, morphological and magnetic behavior of ZnxCo1-xFe2O4/SiO2 nanocomposites. J Alloys Compd. 2019;792:432–43.
Dippong T, Goga F, Levei E-A, Cadar O. Influence of zinc substitution with cobalt on thermal behavior, structure and morphology of zinc ferrite embedded in silica matrix. J Solid State Chem. 2019;275:159–66.
Goto H, Maeda I, Kihara U, Torii M. Single crystal of Mn-substituted NiZn ferrite grown by the Bridgman Method. IEEE Trans Magn. 1983;19(3):1463–6.
Babbar VK, Puri RK. Hot-pressed Mn-Zn-Ni and Mn-Zn-Co ferrites for magnetic recording heads. IEEE Trans Magn. 1992;28:21–6.
Bara JJ, Bogacz BF, Danilkiewicz MI. Mössbauer effect study of the MnxZnyNizFe2O4. Acta Magnetica Sup. 1984;84:271–2.
Zhong H, Zhang H. Effects of different sintering temperature and Mn content on magnetic properties of NiZn ferrites. J Magn Magn Mater. 2004;283:247–50.
Sattar AA, El-Sayed HM, El-Shokrofy KM, El-Tabey MM. Effect of manganese substitution on the magnetic properties of nickel-zinc ferrite. J Mater Eng Perform. 2005;14:99–103.
Sattar AA, El-Sayed HM, El-Shokrofy KM, El-Tabey MM. Study of the dc resistivity and thermoelectric power in Mn-substituted Ni–Zn ferrites. J Mater Sci. 2007;42:149–55.
Rao BP, Kim C-O, Kim C-G, Caltun OF. X-ray and magnetic properties of manganese substituted Ni-Zn ferrites. J Optoelectron Adv Mater. 2007;9:1143–6.
Hossain AKMA, Biswas TS, Mahmud ST, Yanagida T, Tanaka H, Kawai T. Enhancement of initial permeability due to Mn substitution in polycrystalline Ni0.50-xMnxZn0.50Fe2O4. J Magn Magn Mater. 2009;321:81–7.
Singh AK, Verma A, Thakur OP, Prakash C, Goel TC, Mendiratta RG. Electrical and magnetic properties of Mn–Ni–Zn ferrites processed by citrate precursor method. Mater Lett. 2003;57:1040–4.
Singh AK, Verma A, Thakur OP, Prakash C, Goel TC, Mendiratta RG. DC resistivity of Mn–Ni–Zn ferrites. Jpn J Appl Phys. 2002;41:5142–4.
Singh AK, Goel TC, Mendiratta RG, Thakur OP, Prakash C. Dielectric properties of Mn-substituted Ni–Zn ferrites. J Appl Phys. 2002;91:6626–9.
Singh AK, Goel TC, Mendiratta RG, Thakur OP, Prakash C. Magnetic properties of Mn-substituted Ni–Zn ferrites. J Appl Phys. 2002;92:3872–6.
Singh AK, Singh AK, Goel TC, Mendiratta RG. High performance Ni-substituted Mn–Zn ferrites processed by soft chemical technique. J Magn Magn Mater. 2004;281:276–80.
Singh AK, Goel TC, Mendiratta RG. Effect of manganese impurity on the conductivity, dielectric behavior and magnetic properties of Ni0.3MnxZn0.7-xFe2O4. Jpn J Appl Phys. 2003;42:2690.
Singh AK, Goel TC, Mendiratta RG. Effect of cation distribution on the properties of Mn0.2ZnxNi0.8-xFe2O4. Solid State Commun. 2003;125:121–5.
Singh AK, Goel TC, Mendiratta RG. Low-temperature synthesis of Mn0.2Ni0.2Zn0.6Fe2O4 ferrites by citrate precursor method and study of their properties. Phys Stat Sol (A). 2004;201:1453–7.
Verma A, Chatterjee R. Effect of zinc concentration on the structural, electrical and magnetic properties of mixed Mn–Zn and Ni–Zn ferrites synthesized by the citrate precursor technique. J Magn Magn Mater. 2006;306:313–20.
Bueno AR, Gregori ML, Nóbrega MCS. Microwave-absorbing properties of Ni0.50-xZn0.50-xMe2xFe2O4 (Me = Cu, Mn, Mg) ferrite—wax composite in X-band frequencies. J Magn Magn Mater. 2008;320:864–70.
Bueno AR, Gregori ML, Nóbrega MCS. Effect of Mn substitution on the microstructure and magnetic properties of Ni0.50-xZn0.50-xMn2xFe2O4 ferrite prepared by the citrate–nitrate precursor method. Mater Chem Phys. 2007;105:229–33.
Mathur P, Thakur A, Singh M, Harris G. Preparation and characterization of Mn0.4NixZn0.6-xFe2O4 soft spinel ferrites for low and high frequency applications by citrate precursor method. Z Phys Chem. 2008;222:621–33.
Thota S, Kashyap SC, Sharma SK, Reddy VR. Cation distribution in Ni-substituted Mn0.5Zn0.5Fe2O4 nanoparticles: a Raman, Mössbauer, X-ray diffraction and electron spectroscopy study. Mater Sci Eng B. 2016;206:69–78.
Shirsath SE, Toksha BG, Kadam RH, Patange SM, Mane DR, Jangam GS, Ghasemi A. Doping effect of Mn2+ on the magnetic behavior in Ni–Zn ferrite nanoparticles prepared by sol–gel auto-combustion. J Phys Chem Solids. 2010;71:1669–75.
Mane DR, Birajdar DD, Shirsath SE, Telugu RA, Kadam RH. Structural and magnetic characterizations of Mn-Ni-Zn ferrite nanoparticles. Phys Status Solidi A. 2010;207:2355–63.
Xi G, Xi Y, Xu H, Wang L. Study of the preparation of NI–Mn–Zn ferrite using spent NI–MH and alkaline Zn–Mn batteries. J Magn Magn Mater. 2016;398:196–9.
Ramesh S, Sekhar BC, Rao PSVS. Rao BP microstructural and magnetic behavior of mixed Ni-Zn-Co and Ni-Zn-Mn ferrites. Ceram Int. 2014;40:8729–35.
Ramesh S, Dhanalakshmi B, Sekhar BC, Rao PSVS, Rao BP. Structural and magnetic studies on Mn-doped Ni–Zn ferrite nanoparticles. Appl Phys A. 2016;122:984.
Jalaiah K, Babu KV. Structural, magnetic and electrical properties of nickel doped Mn-Zn spinel ferrite synthesized by sol-gel method. J Magn Magn Mater. 2017;423:275–80.
Venkataraju C, Sathishkumar G, Sivakumar K. Effect of cation distribution on the structural and magnetic properties of nickel substituted nanosized Mn–Zn ferrites prepared by co-precipitation method. J Magn Magn Mater. 2010;322:230–3.
Gawas UB, Verenkar VMS, Patil DR. nanostructured ferrite based electronic nose sensitive to ammonia at room temperature. Sens Transducers. 2011;134:45–55.
Gawas UB, Verenkar VMS. Synthesis, thermo-analytical and IR spectral studies of hydrazinated mixed metal carboxylates : a single source precursor to nanosize mixed metal oxides. Thermochim Acta. 2013;556:41–6.
Sawant SY, Kannan KR, Verenkar VMS. Synthesis, characterization and thermal analysis of nickel manganese fumarato-hydrazinate. In: Proceeding of the 13th National symposium on thermal analysis, B.A.R.C. Mumbai: Indian Thermal Analysis Society, 2002, pp. 154–155.
Rane KS, Verenkar VMS. Synthesis of ferrite grade γ-Fe2O3. Bull Mater Sci. 2001;24:39–45.
Gonsalves LR, Verenkar VMS, Mojumdar SC. Preparation and characterization of Co0.5Zn0.5Fe2(C4H2O4)3·6N2H4: a precursor to prepare Co0.5Zn0.5Fe2O4 nanoparticles. J Therm Anal Calorim. 2009;96:53–7.
Coutinho DM. Verenkar VMS Preparation, spectroscopic and thermal analysis of hexahydrazine nickel cobalt ferrous succinate precursor and study of solid-state properties of its nanosized thermal product Ni0.5Co0.5Fe2O4. J Therm Anal Calorim. 2017;128:807–17.
Dessai PPG, Verenkar VMS. Structural, electrical and magnetic properties of nanosize and bulk Ni0.7Zn0.3Fe2O4 obtained by thermal autocatalytic decomposition of Ni0.7Zn0.3Fe2(C4H2O4)3·6N2H4 precursor. J Mater Sci Mater Electron. 2018;29:6924–36.
Vogel’s text book of quantitative inorganic analysis (Revised by G. H. Jeffery, J. Bassett, J. Mendham and R. C. Danney) 5th edition, Logman, UK (1989).
Wilkins DH. The determination of Nickel, Cobalt, Iron and Zinc in ferrites. Anal Chim Acta. 1959;20:271–3.
Kraus KA, Moore GE. Anion Exchange Studies. VI. 1,2 The divalent transition elements Manganese to Zinc in Hydrochloric Acid. J Am Chem Soc. 1953;75:1460–2.
Acknowledgement
The authors are grateful for the financial support from DST, New Delhi through the Nano Mission project No. SR/NM/NS-86/2009 and also under FIST. The authors are also thankful to UGC, New Delhi, for the financial support under SAP. One of the authors, Prajyoti P. Gauns Dessai, would like to acknowledge DST, New Delhi, for providing financial support in the form of DST-INSPIRE fellowship. The authors are also thankful to Mr. Girish Prabhu, NIO-Goa for XRD facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gauns Dessai, P.P., Verenkar, V.M.S. Synthesis and characterization of Ni0.7−xMnxZn0.3Fe2(C4H2O4)3·6N2H4 (x = 0.1–0.6): A precursor for the synthesis of nickel–manganese–zinc ferrites. J Therm Anal Calorim 142, 1399–1411 (2020). https://doi.org/10.1007/s10973-020-09485-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09485-2