
Abstract
In this study, CaCl2- and MgCl2–alcohol solvates of different stoichiometric quantities of ethyl alcohol (EtOH) and methyl alcohol (MeOH) were synthesized and characterized via coupled thermogravimetric–differential scanning calorimetry, thermogravimetric–mass spectrometric evolved gas analysis (TG-MS), spectroscopic analysis (Raman) methods as well as by X-ray diffraction. Correlations between the obtained calorimetric, thermodynamic, kinetic, and crystallographic data were carried out. The CaCl2–alcohol systems seem suitable for heat storage based on the feasible recovery of the salt. However, Raman spectroscopic results revealed that the MgCl2–EtOH solvates were instable compounds. Irreversible transformation of MgCl2–alcohol solvates related to the formation of alkyl chloride appeared upon heating, as proven by TG-MS and Raman spectroscopic measurements. Pure salt–alcohol solvates could not be prepared under technically applicable conditions. The samples contained at least traces of water. Appearance of side reactions resulting in magnesium oxychlorides, oxyhydroxides, and possible release of HCl with cycling may contribute to corrosion of reactor components. Based on these considerations, MgCl2–alcohol solvate systems are not recommended for heat storage.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Storage and utilization of thermal energy are of utmost importance for a sustainable energy future. There are three main approaches in this field, e.g., sensible heat storage in water [1, 2], utilization of latent heat by phase change materials (PCM) [3,4,5,6,7,8,9,10,11,12,13,14,15], and conversion to chemical energy by different thermochemical reactions [16,17,18,19,20,21,22]. Heat storage via reversible thermochemical reactions has several benefits, such as high energy density per volume material and low long-term losses. Space can be a limiting factor for many practical applications. Most mobile applications, for instance, demand high energy densities in small volumes.
To date, the reaction of inorganic salts with water to form salt hydrates under heat release (1) has mainly been studied and optimized [23,24,25,26]. Alcohols are known to undergo similar reactions with salts (2). These reactions have only been studied in a casual manner in terms of their application in thermal energy storage [27, 28], although the high vapor pressure and low freezing point of EtOH and MeOH facilitate low-temperature applications unlike the aqueous adducts.
During the exothermic association reaction (discharging mode), the solid inorganic salt MX reacts with the gaseous alcohol R-OH forming the salt–alcohol solvate (alcoholate) MX·zR-OH, where z is the stoichiometric coefficient. The chemical energy stored in the reaction is released as heat of reaction ΔrH2. The reverse reaction (charging mode) is endothermic. Heat energy must be supplied to initiate the dissociation of the MX·zR-OH adduct. The gaseous product R-OH can be collected and condensed. Therefore, the storage volume can be reduced and the reaction products can be stored separately. As a result, a long-term storage without sensible heat losses is technically feasible. A schematic diagram of the operating principle of a closed thermochemical heat storage system is shown in Fig. 1.

Schematic diagram of the operating principle of a closed thermochemical heat storage system
Fast reaction kinetics, precise thermodynamic control, and low regeneration temperatures make salt–alcohol pairs an advantageous option. Recently, the suitability of EtOH solvates of CaCl2, MgCl2, and their mixtures as heat storage materials for practical implementations has been studied [29]. The EtOH sorption ability of CaCl2 was better than that of MgCl2. At high EtOH vapor pressures, overstoichiometric EtOH uptake occurred. The CaCl2–EtOH reaction system exhibited convenient sorption properties coupled with good multi-cyclic stability. Consequently, it has a great potential for low-grade thermal energy storage. However, poor reversibility appeared in case of the MgCl2–EtOH system with increasing number of cycles, probably caused by irreversible decomposition that strongly reduced the material’s sorption performance [29].
Due to the variety of synthesis procedures and conditions, an apparent discrepancy of the stoichiometry of salt alcoholates is found in the published literature. Moreover, present work on the energy storage density is limited. The aim of this work was to prepare and characterize salt–alcohol solvates of different stoichiometry based on anhydrous CaCl2 and MgCl2 by use of commercially available, specially non-purified chemicals with methods that can be relatively easily transposed into practice. The salt–alcohol solvates were prepared by direct synthesis from CaCl2/MgCl2 and EtOH/MeOH. This preparation method is based on the operating principle of a thermochemical heat store and can easily be adapted to practice. The focus was laid not only on the detailed comparative study of the calorimetric behavior, but also on the mapping of the heat-induced changes in these solvates. Besides coupled thermogravimetric–mass spectrometric (TG-MS) [30, 31], thermogravimetric analysis/differential scanning calorimetric (TGA/DSC) [32] techniques are generally applied in heat storage studies. We believed that Raman spectroscopy [33, 34] and X-ray powder diffraction (XRD) [35] methods are informative in sample characterization. Use of above techniques on structural changes in salt alcoholates is quite unique. We tried to find relationships between structural changes in the salt–alcohol solvates and the cyclic stability during repeated alcoholation/dealcoholation reactions observed earlier [29].
In solid state, the solvent molecules could be in a well-localized form and in solution they could be located in the first, second, and so on solvation shell. The interaction energies between the solvent molecules and solvated ions can vary over a broad range. Conducting single-crystal diffraction experiments for each composition was not possible and may be the subject of another work. Throughout this article, the term built-in form refers to the solvent molecules coordinated to the salt ions and also to the molecules loosely bound to the complexes studied, which dissociate upon crystallization or slightly elevating the temperature.
Experimental
Synthesis of salt–alcohol solvates
Anhydrous CaCl2 powder (Merck, Ph Eur), anhydrous MgCl2 powder (Roth, ≥ 98.5%), absolute MeOH (max. 0.003% H2O, Merck), and absolute EtOH (max. 0.01% H2O, Merck) were used as starting materials. The H2O content of pure CaCl2 and MgCl2 samples as received was determined by TGA. The powdered salts contained only traces of H2O.
Alcohol-salt pairs in four combinations, i.e., MeOH/CaCl2, EtOH/CaCl2, MeOH/MgCl2, EtOH/MgCl2, were prepared setting different alcohol/salt molar ratios (Table 1). The alcohol/salt stoichiometric ratios varied between 2 and 8 mol of EtOH or MeOH per mole anhydrous CaCl2 or MgCl2. Samples of 10 g of the respective salt were placed into a round bottom flask that was purged with dry nitrogen (N2) prior to each alcoholate synthesis reaction. The flask was equipped with a magnetic stirrer and two thermometers. A stoichiometric amount of liquid alcohol was dropped onto the vigorously agitated salt under dry N2 atmosphere, while both the temperature of the sample and the vapor phase were measured. Changes in textural and optical properties of the alcoholated salts were recorded. Preparation and denomination of the samples obtained are summarized in Table 1. The prepared samples were stored in closed glass vessels in a vacuum desiccator over P2O5.
Characterization methods
Simultaneous thermogravimetric analysis and differential scanning calorimetry
Experimental alcohol/salt ratios of the salt–alcohol solvates prepared at different theoretical stoichiometric molar ratios, according to Table 1, and associated heat absorptions were identified by simultaneous TGA/DSC. The horizontal furnace TGA/DSC 1 from Mettler Toledo was used. Specimens of 10–15 mg were uniformly placed into alumina crucibles of 70 µL volume and kept isothermally at 30 °C for 15 min to stabilize the sample. Dynamic runs were carried out by scanning the specimen from 30 to 180 °C with a temperature ramp of 3 °C min−1 at atmospheric pressure. During subsequent isothermal stabilization at the final temperature, the mass was recorded until the reaction went to completion. The TGA/DSC instrument was purged with N2 at a flow rate of 50 mL min−1 during the entire measurement. Effects of buoyancy forces and temperature changes on the TGA and DSC signal were eliminated by automatic blank curve correction. For subtraction of the blank curve from the measurement curve, a blank curve was recorded under the same temperature conditions as the measurement curve, but using empty reference and sample crucibles. All measurements were performed under well-controlled laboratory conditions. The TGA/DSC device was calibrated with high purity metal standards (gallium, indium, lead, aluminum, and gold) over the temperature range of interest. For temperature and enthalpy calibration, the calibration substances were subjected to the same temperature and heating conditions as the samples analyzed. The measured onset temperature, which is assigned the start of the melting process, was compared with the reference melting point of the respective standard. The determined enthalpy of fusion was also validated by comparison with the reference value. The blank curve reproducibility was better than ± 10 µg over the whole temperature range. The standard deviation of the enthalpy reproducibility was given as < 5%. Mass changes and heat powers were determined with a precision of ± 0.1 µg and ± 1 mW. The Mettler Toledo STARe software 11.00a was used for data processing.
Selected samples of the series of salt–alcohol solvates were characterized complimentary by Raman spectroscopy, TG-MS, and XRD. In case of the first two methods, N2 atmosphere was used for sample handling.
Raman spectroscopy
Raman spectra of samples in closed glass ampoules held at room temperature were recorded with a dynamically aligned Bio-Rad (Digilab) dedicated FT-Raman spectrometer equipped with a Spectra Physics Nd–YAG laser (1064 nm) and high sensitivity liquid-N2-cooled Ge detector. The excitation laser power used was about 250 mW at the samples. Increase in laser power up to 500 mW does not result in a significant change of the spectra that indicates the stability of the samples during the measurement. The resolution of the Raman instrument was ca. 4 cm−1, and a backscattered geometry was used. For each spectrum, 256 individual spectra were averaged.
Thermogravimetric–mass spectrometric evolved gas analysis
The simultaneous thermogravimetric and mass spectrometric evolved gas analyses (TG-MS) were recorded on a Setaram LabsysEvo thermal analyzer, in high purity (99.9999%) helium atmosphere, with a flow rate of 80 mL min−1. The measurements were done with a heating rate of 20 °C min−1; the samples were weighed into 100 μL aluminum crucibles in inert (dry N2) atmosphere. Exposure to moisture during sample transfer was prevented, and MS recording of evolved volatiles was enabled by closing the crucibles with aluminum lids pierced with a 700 micron hole by crimping. The measurements were performed in the 25–500 °C temperature range. The obtained results were baseline corrected and then evaluated with the thermal analyzer’s processing software (AKTS Calisto Processing, ver. 1.41). Parallel with the thermogravimetric measurements, the analysis of the evolved gases/volatiles was carried out on a Pfeiffer Vacuum OmniStar™ gas analysis system coupled to the above-described TGA. The gas splitter and transfer line to the mass spectrometer were preheated to 250 °C. The scanned m/z interval was 5–79 amu, with a scan speed of 20 ms amu−1. The mass spectrometer was operated in electron impact mode. During a measurement, the total ion current (TIC), the discrete ion current of all scanned masses (75 masses) and the analog spectra on each scan cycle (1 scan cycle was 1.5 s long) were obtained in parallel.
X-ray powder diffraction
XRD patterns were obtained in a Philips model PW 3710 based PW 1050 Bragg–Brentano parafocusing goniometer using CuKα radiation (λ = 0.15418 nm), graphite monochromator and proportional counter. Samples were placed in an Anton-Paar HTK2000 high-temperature oven chamber. Prior to measurements, the chamber was flushed with high purity N2 (99.9999%) and during XRD measurements a continuous slow N2 flow was ensured. The temperature steps chosen for the “temperature programmed” XRD were based on the results of the TG-MS measurement of the appropriate sample. First, XRD patterns were recorded at room temperature, which was followed by stepwise in situ heating. The heating rate was 5 °C min−1. XRD patterns were recorded at every temperature step. Finally, the samples were allowed to cool down (20 °C min−1) and the room temperature XRD pattern was again measured. For the phase analysis, reference cards from the ICDD PDF-4 (2010) database were used.
Results and discussion
Preliminary results of the direct synthesis of salt–alcohol solvate
During the formation of the different salt alcoholate solvates, heat was evolved and resulted in a change in the temperature. The temperature change in the sample and the surrounding vapor phase was calculated from the difference between the respective initial and final temperatures. The initial sample temperatures ranged from 24 to 27 °C. The increase in temperature in both the sample and vapor chamber laid in the interval of 15–25 °C for almost all salt–alcohol pairs (Fig. 2). Rather fast reactions occurred and maximum temperatures of 40–56 °C were reached within a time of less than 3 min. In general, the temperature rise was higher in the solid phase than in the vapor phase, except for some outliers. For CaCl2–alcohol solvates, higher temperature differences were measured when compared with that for MgCl2–alcohol solvates. Since the temperature difference of both CaCl2–MeOH and CaCl2–EtOH solvates varied between 20 °C and 25 °C, irrespective of the alcohol/salt molar ratio, similar stoichiometric compositions of the different salt–alcohol solvates were considered. It is assumed that some heat dissipated into the ambient.

Temperature increase during the synthesis of salt–alcohol solvates, according to Table 1. MeOH methanol; EtOH ethanol; diamond—sample, square—vapor chamber
Lower temperature differences compared with that of CaCl2 hydrates [36, 37] indicate lower heat outputs and corresponding enthalpies of reaction. Heterogeneous compounds were observed particularly for samples of higher stoichiometry. Some compositions consisted of two phases: a solid salt alcoholate phase and an excess liquid alcohol phase containing dissolved salt particles. Thus, further analysis was carried out using the solid salt–alcohol solvates only.
Results of TGA/DSC measurements of all samples
The composition of the different salt–alcohol solvates synthesized according to Table 1 was analyzed by TGA/DSC technique. The respective alcohol/salt molar ratios were derived from the change in sample mass recorded as a function of both time and temperature.
Thermal analysis of the CaCl2–MeOH system
The total amount of MeOH uptake of CaCl2 was calculated to be 4 (Table 2). Samples decomposed in 2–3 overlapping stages over a temperature domain of 33–145 °C (Fig. 3). The majority of MeOH was given off below 100 °C. Peak temperatures of 57–94 °C, 80–114 °C, and 95–118 °C were measured (Table 2). As depicted in Fig. 3, the mass loss rate of the first decomposition step, also referred to as rate of dealcoholation, increased with increasing stoichiometric ratio despite similar sample masses. At theoretical alcohol/salt ratios of 2 and 3, a compound doubly coordinated with MeOH was the only CaCl2–MeOH solvate found. This result is consistent with the findings of other authors [27, 38,39,40,41,42]. With excess of absolute MeOH, even a CaCl2 trimethanolate and CaCl2 tetramethanolate could be synthesized (Table 2). Note that samples prepared at higher theoretical alcohol/salt ratios were a heterogeneous mixture of two phases: a solid phase and a liquid phase. The latter was discarded from analysis. The formation of CaCl2–MeOH complexes of different stoichiometry has been claimed by other researchers. Gmelin [43] has reported the existence of CaCl2 monomethanolates and CaCl2 trimethanolates that have been identified in a study conducted by Gerhold and Kahovec. Bonnell [44] and Menschutkin [45] have obtained CaCl2 trimethanolates, too. MeOH solvates of CaCl2 with a molar ratio of 4 [43,44,45,46,47,48] and 6 [49] have been characterized by different analysis methods. The standard reaction enthalpies were deduced from the DSC curve by peak integration and varied between 101 and 151 kJ mol−1 (Table 2).

Mass (TGA) and mass loss rate (DTG) curves of the dealcoholation of CaCl2–MeOH solvates prepared in neat at different MeOH/CaCl2 molar ratios (see Table 1), (measurement temperature interval: 30–180 °C; heating rate of 3 °C min−1; 20 min isotherm at 180 °C)
Thermal analysis of the CaCl2–EtOH system
In CaCl2–EtOH solvate complexes (Fig. 4), the EtOH was evolved in up to four inseparable steps with peak temperature around 68–72 °C, 87–92 °C, 108–121 °C, and 150–177 °C, respectively (Table 2). The decomposition started roughly at 33 °C and was completed at around 178 °C. The major amount of EtOH was desorbed below 100 °C. The rate of deethanolation varied over a broad range and was not reproducible, as shown in Fig. 4. The calculated percentage of mass loss (Table 2) clearly showed that in each sample only 2 mol of EtOH was adsorbed per mole of anhydrous CaCl2. However, a variation in the standard enthalpy of reaction with values between 92 and 130 kJ mol−1 was observed. We suppose that CaCl2 could only hold up to two molecules of EtOH under the experimental conditions studied, irrespective of the initial stoichiometric ratio we used during sample preparation. These data are consistent with the findings described in the literature [27, 50]. According to published literature data, EtOH forms also a number of other CaCl2 ethanolates. Monoethanolates [43, 50, 51], triethanolates [43,44,45, 52, 53], and tetraethanolates [53,54,55] of CaCl2 have been reported previously. Their existence was not proven in this study.

Mass (TGA) and mass loss rate (DTG) curves of the dealcoholation of CaCl2–EtOH solvates prepared in neat at different EtOH/CaCl2 molar ratios (see Table 1), (measurement temperature interval: 30–180 °C; heating rate of 3 °C min−1; 20 min isotherm at 180 °C)
Thermal analysis of the MgCl2–MeOH system
The experimental alcohol/salt ratios of the MgCl2 methanolates prepared, as listed in Table 1, varied significantly from the theoretical ones. Methanolates of lower methanolation states were formed when the absolute MeOH was added in excess under the conditions studied. According to the obtained experimental data, about 1, 2, 3, and 4 MeOH molecules were sorbed by one molecule of MgCl2 (Table 2). Samples 13 and 14 were deliquescent and therefore we presume that both physically and chemically attached MeOH was present. Depending on the amount of MeOH, the dissociation reaction proceeded in 3–4 incomplete steps (Fig. 5) over the temperature region of 33–177 °C with peak temperatures of 65–94 °C, 76–103 °C, 120–136 °C, and 158 °C, respectively (Table 2). Trace amounts of MeOH continued to evolve during isothermal stabilization at 180 °C until the demethanolation went to completion. As plotted in Fig. 5, the mass loss rates are in general lower compared with that of the CaCl2–MeOH and CaCl2–EtOH solvates. The DSC peak integration yielded standard reaction enthalpy values of 69–183 kJ mol−1 (Table 2). The obtained values were raised by increasing the alcohol/salt molar ratio. Recently, MgCl2 monomethanolates have only been reported by Iyimen-Schwarz [51]. Furthermore, MgCl2–MeOH species with stoichiometric numbers of coordinated MeOH molecules of 2 [42, 51, 56], 3 [51, 57], 4 [28, 42, 56, 58, 59], and 6 [47, 49, 56, 57, 60,61,62,63,64,65,66] have been identified in the literature.

Mass (TGA) and mass loss rate (DTG) curves of the dealcoholation of MgCl2–MeOH solvates prepared in neat at different MeOH/MgCl2 molar ratios (Table 1), (measurement temperature interval: 30–180 °C; heating rate of 3 °C min−1; 20 min isotherm at 180 °C)
Thermal analysis of the MgCl2–EtOH system
MgCl2 formed ethanolates of different stoichiometry: stoichiometric ratios of 1.9, 2.3, 3.5, and 4.6 were identified under the experimental conditions studied (Table 2). The latter MgCl2–4.6EtOH is doubtable as the maximum quantity of absolute EtOH added to anhydrous MgCl2 was 4. The dissociation reaction occurred in 2–4 steps (Fig. 6) between 33 and 177 °C with peak temperatures at 61–67 °C, 101–140 °C, and 136–153 °C (Table 2). Trace amounts of EtOH were desorbed during the subsequent isothermal stabilization segment until the reaction went to completion, as observed for MgCl2 methanolates (Fig. 5). The EtOH released in a stepwise manner was probably uncoordinated and coordinated EtOH. The samples 17 and 18 consisted of a two phase mixtures: liquid and solid phase. The latter was used for characterization studies. Mass loss rates between 0.2 and 0.4 mg min−1 were measured (Fig. 6). The calculated standard reaction enthalpies amounted to 65–158 kJ mol−1 (Table 2); the standard reaction enthalpies per mole EtOH were surprisingly low (30–45 kJ mol−1), partly lower than the standard enthalpy of vaporization of EtOH (41.680 kJ mol−1) [67]. MgCl2–EtOH solvates have gained considerable interest among researchers for the synthesis of MgCl2-supported Ziegler–Natta catalysts and subsequently a plethora of the literature exists. Besides well-defined compounds, addition compounds of non-integer stoichiometry have also been identified in several studies. Iyimen-Schwarz [51], Tewell et al. [68], Chadwick and Severn [69], and Bart and Roovers [70] have prepared MgCl2 ethanolates with a molar ratio of 0.47, 1, 1.1, and 1.25. The existence of MgCl2 diethanolates has been proven by Multani [56] and Di Noto et al. [71]. Furthermore, EtOH solvates of MgCl2 with molar ratios of 1.5 [72], 1.67 [68, 70], 2.05 and 2.1 [68, 69], 2.5 [70, 73], 2.8 [69, 72], and 3.33 [28, 70, 72] have been synthesized and investigated in detail. Complex compounds with higher stoichiometry of 4, 4.5, and 5 have been obtained by Bart and Roovers [70], Multani [56], and Tewell et al. [68]. The maximum coordination number observed in MgCl2–EtOH complexes has been 6 according to various authors [56, 57, 63, 65,66,67, 70,71,72, 74, 75].

Mass (TGA) and mass loss rate (DTG) curves of the dealcoholation of MgCl2–EtOH solvates prepared in neat at different EtOH/MgCl2 molar ratios (Table 1), (measurement temperature interval: 30–180 °C; heating rate of 3 °C min−1; 20 min isotherm at 180 °C)
Comparing the decompositions temperatures of CaCl2 alcohol solvates indicates that obviously MeOH was stronger associated with the metal chloride than EtOH. For instance, the EtOH evolution from CaCl2–EtOH solvates started at lower temperatures than the desorption of MeOH from CaCl2–MeOH solvates; the last MeOH molecule was liberated at higher temperatures than the last EtOH molecule. This means that during thermal cycling, EtOH can be liberated with lower energy than MeOH. Analogous reaction behavior was observed for MgCl2 alcohol solvates. Moreover, MgCl2–alcohol solvates showed lower dealcoholation rates and thus slower alcoholation/dealcoholation reactions are assumed in comparison with CaCl2 alcohol solvates.
Energy analysis
Variation in the coordination numbers of the salt–alcohol solvates cited in the references and this study could be the result of the diversity in the synthesis procedures and conditions. According to Bart and Roovers [70], the synthesis method plays a crucial role with regard to the nature of the reactants. Salt–alcohol solvates can be prepared by direct synthesis, solution crystallization and elimination of excess solvent, or recrystallization, for example. The alcohol coordinates to the alkaline earth metal cations to form complexes with variable stoichiometry and structural properties that exist either in the solid state or in solution.
Note that the calculated enthalpies of reaction and dissociation (Table 2) tend to be inaccurate as no quantitative differentiation between alcohol molecules and possible H2O molecules coordinated to one molecule of salt could be made at this stage. Samples contained probably trace amounts of H2O that were neglected, since the freshly prepared samples were immediately characterized via TGA/DSC.
A linear regression analysis of the experimentally obtained standard enthalpies of dissociation as a function of the number of alcohol molecules revealed that the applied model fitted well the data (Fig. 7). The coefficient of determination was close to one for each reaction system. Evidently, the standard enthalpy of dissociation increased along the homologous series of the alcohols studied. EtOH solvates possessed higher enthalpies of formation compared to MeOH solvates. The Ca2+ ion is of higher charge than the Mg2+ ion and has a greater polarizing power. Therefore, the standard enthalpies of formation of CaCl2–alcohol complexes are comparatively higher.

Experimentally obtained standard enthalpies of dissociation of various salt–alcohol solvates plotted against the number of alcohol molecules evolved during the dissociation reaction, MeOH methanol, EtOH ethanol. Linear regression analysis was applied to determine the coefficient of determination (R-squared)
The best fit (R2 = 0.9981) was achieved with the CaCl2–EtOH reaction system confirming its chemical stability. The calculated enthalpies of dissociation for all CaCl2–2EtOH solvates analyzed were in the range of 1303–1394 kJ mol−1 (Table 2) and coincided with formation enthalpy and dissociation enthalpy data given in the published literature [27, 50] (Table 3). CaCl2–EtOH complexes of higher ethanolated states as reported by Parker et al. [53] and those of lower ethanolated states [50, 51] could not be synthesized under the experimental conditions applied.
The CaCl2–MeOH system exhibited an acceptable fit with a coefficient of determination of R2 = 0.9967. The enthalpies of dissociation of the CaCl2–2MeOH solvates with values of 1314 kJ mol−1 and 1337 kJ mol−1, respectively, (Table 2) were slightly higher than literature values [27, 38, 42, 51] (Table 3). Enthalpies of reaction and formation/dissociation for CaCl2–3MeOH and CaCl2–4MeOH have not been determined yet. The enthalpy of dissociation of CaCl2–3MeOH and of CaCl2–4MeOH was estimated at 1520 kJ mol−1 and 1771 kJ mol−1/1754 kJ mol−1, respectively. The enthalpy of dissociation of CaCl2–MeOH complexes increased with the number of alcohol molecules indicating a linear relationship. However, a nonlinear relationship could be observed when the standard enthalpies of reaction per mole of alcohol \(\frac{{\Delta_{\text{r}} H^{0} }}{z}\) were compared (Table 2). At levels of methanolation higher than 2, the enthalpies of reaction per mole MeOH varied between 31 and 38 kJ mol−1 (Table 2), which are close to the standard enthalpy of vaporization of MeOH (37.965 kJ mol−1) [67]. Apparently, up to 2 molecules could be chemically bound as a ligand. In MeOH solvates of CaCl2 with higher molar ratios, the inclusion of MeOH molecules or physical incorporation of molecules into the crystal lattice could have caused the decrease in the standard enthalpy of reaction per mole of MeOH.
A linear relation between the enthalpy of dissociation and the number of alcohol molecules was also found for MgCl2–EtOH and MgCl2–MeOH solvate complexes (Fig. 7). The R-squared values of the MgCl2–EtOH and MgCl2–MeOH systems were R2 = 0.9979 and R2 = 0.9971, respectively. Reference data are rarely available. Iyimen-Schwarz [51] has collected the enthalpies of formation and dissociation of MgCl2–1EtOH, MgCl2–1MeOH, and MgCl2–3MeOH solvates from thermal cycling tests by DSC technique (Table 4). When compared with values determined in this study, a significant deviation appeared. The values given by Iyimen-Schwarz [51] are mean values averaged over 13 cycles for MgCl2–MeOH solvates and 10 cycles for MgCl2–EtOH solvates. Maximum formation and dissociation enthalpy values of 31 kJ mol−1/− 34 kJ mol−1 and 111 kJ mol−1/− 103 kJ mol−1 were derived for MgCl2–1MeOH and MgCl2–3MeOH. In the present study, the enthalpies of dissociation of MgCl2–1MeOH, MgCl2–2MeOH, MgCl2–3MeOH, and MgCl2–4MeOH ranged from 891 to 1639 kJ mol−1 (Table 2). The calculated enthalpies of reaction per mole MeOH of 43–77 kJ mol−1 (Table 2) were higher than the standard enthalpy of vaporization of MeOH. However, the enthalpies of reaction per mole EtOH with values of 30–45 kJ mol−1 (Table 2) were lower in comparison with the enthalpy of vaporization of EtOH (42 kJ mol−1) [67]. Iyimen-Schwarz [51] has obtained even lower enthalpies of formation and dissociation and, respectively, enthalpies of reaction per mole of EtOH for MgCl2–EtOH solvates (Table 3). These inconsistent and unreliable experimental results are ascribed to inherent instability issues of the MgCl2–alcohol solvates.
To assess the suitability of the various salt–alcohol solvate systems for low-temperature heat storage, the gravimetric energy density was calculated from the experimentally measured standard enthalpies of reaction. The gravimetric energy density is a thermochemical material characteristic. As a key performance metric, it is used to evaluate and compare the energy storage performance of thermal energy storage systems. The gravimetric energy density of the investigated salt–alcohol systems was similar, irrespective of the reaction pair combination and ranged from 354 to 943 kJ kg−1, respectively. From the energetic point of view, salt–alcohol solvate systems for heat storage are not as good as salt–water solvate systems, as the measured standard enthalpies of reaction and associated gravimetric energy densities are roughly half the respective values of the latter ones [76].
Raman spectroscopic characterization of selected samples
In a starting series of experiments, we increased the laser energy systematically from 200 mW to 500 mW and we found that spectra of sample 6 were not influenced by the change of the excitation energy. This observation let us to conclude that the alcohol solvate sample was stable enough to be characterized by Raman spectroscopy. Raman spectra of several CaCl2–alcohol solvates recorded at room temperature can be seen in Fig. 8. Comparing the spectrum of pure CaCl2, (line d in Fig. 8a) to those of the different alcohol solvates (lines a, b, e, f in Fig. 8), it could be suggested that the very low wavenumber region was characteristic for CaCl2 and bands from ~ 450 to ~ 3100 cm−1 belonged to the alcohols. Although Raman spectroscopy is not actually sensitive to H2O, a very weak band at about 3400 cm−1 indicated that CaCl2 contained a certain amount of coordinated H2O (line e in Fig. 8b). As a comparison, the Raman spectrum of moisture exposed CaCl2 is also shown (line h in Fig. 8b).

Raman spectra of alcohol solvates of CaCl2. a MeOH solvates; b EtOH solvates; a: sample 1; b: sample 4; c: MeOH; d: CaCl2; e: sample 6; f: sample 9; g: EtOH; h: moisture exposed CaCl2
The MeOH in sample 1 (line a in Fig. 8a) was mainly in built-in form as νas of CH3 at 2952 cm−1 and νs of CH3 at 2844 cm−1 and was shifted with ~ 10 cm−1 comparing to those of the MeOH (2942 cm−1 and 2833 cm−1, line c in Fig. 8a). Bands at 3013 cm−1 and 2982 cm−1 in sample 1 indicated the presence of coordinated OCH3 (line a in Fig. 8a), while the band at ~ 3400 cm−1 indicated the presence of certain built-in H2O (cf. line a and line d in Fig. 9a) that was somewhat more intensive than in the starting salt. As the Raman technique gives average information about the material, we could not decide, whether the sample consisted of the mixture of non-coordinated, OH-coordinated, and MeOH-coordinated units of the CaCl2 or units, which had MeOH and H2O in the same coordination sphere.

Raman spectra of MeOH solvates of MgCl2. a overview spectra. b enlargement of CH3 region; a: sample 12; b: sample 13; c: MeOH; d: MgCl2; e: sample 10; f: sample 12; g: sample 13; h: MeOH
Contrary to sample 1, sample 4 contained a large amount of free MeOH besides the coordinated MeOH witnessed by the doublet of νs CH3 band at 2844 cm−1 and 2836 cm−1; the latter belongs to non-coordinated MeOH. In addition, a double or triple band in the region ~ 3300 cm−1 indicated that –OH and non-coordinated MeOH existed in this sample (cf. lines b and c in Fig. 8a). The appearance of a significant amount of non-coordinated MeOH in sample 4 (prepared at an alcohol/salt ratio of 6:1) confirmed that the maximum number of the coordinated MeOH could be 4, as it was already indicated previously (Sect. 3.1). It is worth noting that the Raman spectrum of sample 3 was very similar to that of sample 4, apart from the band of the non-coordinated MeOH, which was somewhat weaker in sample 3 than in sample 4.
Regarding the alcohol solvates obtained from CaCl2 and EtOH, no real difference between the Raman spectra of the samples prepared at different alcohol/salt ratios could be found (cf. line e and f in Fig. 8b) in accordance with the findings described in Sect. 3.1. Certain relative intensity changes in 2973 cm−1/ ~ 2878 cm−1 of νasCH3/νsCH3 in the region of EtOH (cf. lines e, f and g in Fig. 8b) indicated the presence of the EtOH incorporated into the crystal lattice. The shift of the νC-O bands at 1096 cm−1 and 1051 cm−1 to 1088 cm−1 and 1048 cm−1, respectively, could support this idea. However, the appearance of a weak band at 3396 cm−1 confirmed again the presence of coordinated H2O.
Raman spectra of several MgCl2–MeOH solvates are depicted in Fig. 9. Samples in these series were very similar to each other. The splitting of the νC–O band at 1033 cm−1 indicated MeOH incorporation into the crystal structure. The intensity ratios of MeOH bands/Mg–Cl bands increased with the amount of introduced MeOH. The νas of CH3 appeared at 2950 cm−1 and νs of CH3 appeared at 2848 cm−1 in the MgCl2–MeOH solvates. These values were shifted compared to that of free MeOH (cf. lines a, b, c and d in Fig. 9b), which indicated that MeOH definitely existed in built-in form in the samples 10, 12, and 13.
Regarding the MgCl2–EtOH systems (Fig. 10), the spectra resembled the spectrum of EtOH showing only negligible band shifts; it was difficult to decide, whether the EtOH existed in a real built-in form or not (cf. line a and c in Fig. 10a). Surprisingly, a new C-H stretching band at high wavenumber appeared (better visualized for sample 18, and only as a shoulder for sample 15) that might be assigned to νC-H of halogen substituted methyl group. As a comparison, the library IR spectrum of ethyl chloride is also shown (Fig. 10b). It seems plausible that ethyl chloride was formed in MgCl2–EtOH systems. Simultaneously, the Ca–Cl vibrational band at 241 cm−1 almost completely disappeared (cf. line b in Fig. 10a).

Raman spectra of MgCl2–EtOH solvates. a EtOH solvates; b reference FT-IR spectrum of ethyl chloride from Aldrich Vapor FT-IR Spectral Library; a: sample 15; b: sample 18; c: EtOH; d: MgCl2
As a conclusion, MgCl2–EtOH solvates underwent certain decomposition resulting in ethyl chloride formation. Pure salt–alcohol solvates could not be obtained, because of the presence of a certain amount of salt hydrates.
TG-MS behavior of selected samples
As preliminary investigations, the starting materials, i.e., the CaCl2 and MgCl2, were analyzed, and it was found that both of them contained roughly around 2% H2O of crystallization (more precisely, CaCl2 contained 2.28% H2O, while the H2O content of MgCl2 was 2.36%).
Four samples were chosen for the TG-MS measurements, namely 3 (containing CaCl2 and MeOH), 7 (which contain CaCl2 and EtOH), 12 (containing MgCl2 and MeOH), and finally 18 (composed of MgCl2 and EtOH). In Fig. 11, the mass loss and some selected ion currents are plotted against the temperature obtained from sample 3, while on the right side of the figure the chemical species corresponding to the chosen m/z values are also shown. On the TG curve, between 55 and 300.5 °C, three mass loss steps can be seen. The first mass loss step is between 55 and 123.5 °C, with a mass loss of 23.3%, the second step is between 123.5 and 178 °C, with 19.2% mass lost, and the last step between 178 and 300.5 °C resulting in 9.5% mass loss; the total mass loss during the measurement was 52.9%. Comparing the shape of the three ion currents, three overlapping peaks can be observed on each curve, which correspond to the appropriate mass loss step. The m/z 32 is the molecular ion of MeOH, the m/z 31 is the base peak of MeOH (and also a characteristic marker of aliphatic alcohols), while the m/z 15 corresponds to the methyl ion (CH3). From the TG-MS measurement, it can be concluded that the evolution of MeOH starts at very low temperatures (around 30 °C). Some H2O was also detected during the measurement (the ion current of H2O is not shown), but its concentration variation was within one order of magnitude compared to the concentration variation of MeOH, which was 2 orders of magnitude (m/z 31), thus confirming that the major volatile component formed during the measurement was MeOH. Moreover, no chlorinated compounds (e.g., methyl chloride) were detected. It can be seen that above 300 °C all three ion current curves are still decreasing, which implies that some MeOH is still lost, but this causes a very small mass loss, not detected by TG.

TG-MS trace of the sample 3 (mass loss on the upper part, while ion currents on the lower part of the graph), on the right side the formulae of the corresponding fragments/ions
Considering the TG-MS trace of the sample 7 (Fig. 12), only two mass loss steps can be distinguished. The first mass loss step is between 55 and 95 °C, with a mass loss of 1.9%, while the second step is between 95 and 241 °C, with a mass loss of 48%; the total mass loss during the measurement was 50.3%. It can be seen that the formation of EtOH vapors (m/z 46 is the molecular ion of EtOH, m/z 45 is a deprotonated EtOH, while m/z 31 is the base peak of EtOH) begins at very low temperatures, as already observed in the case of sample 3.

TG-MS trace of the sample 7, on the right side the formulae of the corresponding fragments/ions
A very small amount of H2O can be detected between 152 and 235 °C, which means that the H2O is much more strongly bound to the CaCl2 than the EtOH. No alkyl chloride was detected proving the thermal stability of the CaCl2–alcohol solvate. Comparing the results of sample 3 and 7, it can be concluded that EtOH (sample 7) is lost in a narrower temperature range than MeOH (sample 3). Moreover, the temperature value, where the alcohol is practically lost decreases from 300.5 °C (sample 3) to 240 °C (sample 7). This means that during cyclic measurements, EtOH can be liberated with less energy than MeOH.
Replacing CaCl2 with MgCl2, significant differences appear in the TG-MS trace of sample 12 and 18. On the mass loss curve of the sample 12 (Fig. 13), three mass loss steps can be identified. The first larger step is between 55 and 198 °C, with a mass loss of 40%, the second, smaller mass loss is between 198 and 306 °C (10% mass lost), and the last, very small step is from 306 °C up to the end of the measurement (up to 500 °C, 1.2% mass loss). Evaluating the ion current curves of four masses (m/z 32 molecular ion of MeOH, m/z 31 base peak of MeOH, while m/z 52 is the molecular ion of methyl chloride, with the 37Cl isotope, and m/z 50 is the molecular ion of methyl chloride, with the 35Cl isotope), it can be seen that MeOH is liberated below 180 °C. Above this temperature, besides MeOH, which is still the major component of the volatiles and some traces of H2O, the formation of methyl chloride begins. This confirms that at higher temperatures, MgCl2 hydrolyzes and converts the MeOH into methyl chloride. Around the upper end of the temperature scale, a small amount of methyl chloride is still released between 410 and 490 °C (0.7% mass is lost).

TG-MS trace of the sample 12, on the right side the formulae of the corresponding fragments/ions
Replacing the MeOH in the magnesium salt with EtOH (sample 18, Fig. 14), again a complicated, multi-step decomposition pattern is obtained. On the mass loss curve of sample 18, four decomposition steps can be identified (first step between 50 and 100 °C, mass loss 4.2%; second, larger step between 100 and 170 °C, mass loss 46.3%, third step between 170 and 212 °C, mass loss 13.3% and fourth step between 212 and 288 °C, mass loss 10.3%). It can be seen that below 140 °C, the mass loss is caused mainly by the evaporation of EtOH (m/z 46 is the molecular ion of the EtOH, m/z 45 is a deprotonated EtOH, while m/z 31 is the base peak of EtOH) and some traces of H2O (ion curves not shown). Above 140 °C, the formation of ethyl chlorides (m/z 66 is the molecular ion of ethyl chloride, with the 37Cl isotope, while m/z 64 is the molecular ion of ethyl chloride, with the 35Cl isotope) can be detected. Ethyl chloride is formed over a broad temperature range (between 140 and 370 °C), additionally the concentration of ethanol decreases above 290 °C, while the ethyl chloride is still formed. The formation of ethyl chloride confirms the fact that MgCl2 hydrolyzes and converts the EtOH into the corresponding alkyl chloride. Comparing the TG-MS results of sample 12 and 18, it can be seen that the alkyl chloride formation starts at lower temperatures in the case of sample 18 (140 °C). The end temperature value of the mass loss end is shifted toward lower temperatures (mass loss end temperature of sample 12 is 306 °C, while the corresponding value is 288 °C in the case of sample 18).

TG-MS trace of the sample 18, on the right side the formulae of the corresponding fragments/ions
Temperature programmed XRD characterization of selected samples
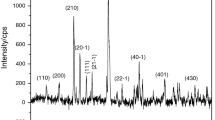
Figure 15 shows the XRD patterns of four selected salt–alcohol solvate samples recorded by use of stepwise heating. The temperature steps were also chosen according to the TG curve obtained from the TG-MS measurement.

XRD patterns of selected alcohol solvates at different temperatures (temperature steps adjusted according to TG curve obtained from the TG-MS measurement; heating rate 5 °C min−1; atmosphere: dry N2). a MeOH–CaCl2 (sample 3); b EtOH–CaCl2 (sample 7); c MeOH–MgCl2 (sample 12); d EtOH/MgCl2 (sample 18)
Sample 3 in its starting form (Fig. 15) consisted of MeOH-coordinated phases. The XRD diffraction pattern was somewhat more complicated at 55 °C than at 25 °C, which indicated the formation of a more complex system by the mild heating. These samples consisted of certain CaCl2xMeOH (H2O) phases. The most likely composition was the average CaCl2/MeOH/H2O = 1:1:3 ratio in the coordination sphere, which could be estimated from TG. Either a mixture of 1:4 and 1:2 coordinated samples of CaCl2–H2O and CaCl2–MeOH or samples with mixed coordination spheres could be imagined. At 120 °C, 177 °C, and 301 °C samples were CaCl2-like materials having amorphous part. At 500 °C, only the CaCl2 phase existed. The sample after re-cooling was CaCl2 with some Ca-hydroxide-carbonate. The XRD patterns at 120 °C, 177 °C, and 301 °C were very similar in spite of the large amount of MeOH that went away in the above temperature steps, which implies that a significant amount of MeOH was not really bound in the coordination sphere of the salt.
The diffraction pattern of sample 7 (Fig. 15b) did not change significantly during the heating up to 53 °C in accordance with the TG-MS results, which shows that no significant amount of EtOH or H2O was removed during the first some minutes. The starting solvate formed at 1:3 molar ratio could be imagined as a mixture of crystals with coordination sphere 1:2 and 1:4 or crystals with coordination sphere 1:2 and alcohol inclusions. The situation was complicated with the presence of H2O. Consequently, besides the pure CaCl2·zEtOH and pure CaCl2·zH2O crystals, CaCl2·(x–y)EtOH·yH2O structures could exist. Based on the XRD results it was impossible to give a more detailed description. Pure CaCl2 phase could also be found in the region of 55–500 °C. Probably easily decomposable alcoholic, a relatively stable alcoholic and hydrous phases existed parallel in this sample. The final state was alcohol- and H2O-free CaCl2.
The composition of the initial sample 12 was rather complicated (Fig. 15c). A volatile part was removed at 56 °C that resulted in a slight change in the diffraction pattern. One might think that physisorbed solvent was removed during the heating from 25 up to 56 °C and the solid consisted of mainly several H2O-coordinated MgCl2 and MeOH-coordinated MgCl2 forms. Regarding the starting solvate, the TG-MS indicated parallel removing of MeOH and a significant amount of H2O. The TG-MS gave a MgCl2/MeOH ratio of 3.12 only instead of 4. Since the molar mass of H2O is smaller than that of MeOH, the difference might come from the presence of structures with coordination number 4, but both ligands were in the coordination sphere in different variation. Consequently, the starting solvate itself had to be a mixture. The slight changes in the XRD pattern of sample 12 at 56 °C compared to that at 25 °C resulted from the small variation of the individual components. At 200 °C, the broadenings in the XRD patterns indicated the formation of amorphous phases. A less crystalline form observed was supposed to be a mixture of MeOH- and H2O-coordinated materials. It is conceivable that MeOH and H2O bound to the salt more or less equally strongly. Some small peaks at about 2θ ~ 32° and 2θ ~ 50° decreased upon increasing the temperature, but we can only say that probably the decomposition of 1:2 complexes appeared. At 307 °C either H2O or MeOH could not be present according to TG-MS. The XRD pattern shows the presence of a certain amount of MgCl2 and/or MgOCl besides amorphous phase(s). At 500 °C, the final material was more crystallized; the final state was MgO (with certain hydrate) and probable Mg-hydroxy-hydrate. The presence of certain MgOOH·HCl could be supposed, too. From these results, it is obvious that the MgCl2 decomposed upon increasing the temperature from 307 to 500 °C. When the sample was cooled to 25 °C, the XRD pattern (and of course the structure) formed at 500 °C was maintained.
The XRD patterns of sample 18 (Fig. 15d) at low temperatures (25 °C, 53 °C) were very similar to those of MgCl2·zH2O. However, TG-MS had shown the removal of mainly EtOH (fragments of EtOH). This observation excluded that the starting material was merely MgCl2.zH2O. We had to assume that MgCl2.zH2O is isostructural with MgCl2·zEtOH. Only a little shift of the peaks appeared in comparison with MgCl2.zH2O, which means that the point group was the same in both cases. Because the shift was small, the cell parameters were not altering significantly. This was very surprising considering the difference in the steric properties of H2O and EtOH. The very small change in the XRD pattern at 53 °C compared to the XRD pattern at 25 °C could be explained by the removal of a small amount of non-chemisorbed alcohol (physisorbed EtOH or inclusion of EtOH). In the temperature region of 101–306 °C, the sample gradually lost the EtOH. According to TG-MS results, only EtOH was removed up to 171 °C; EtOH and a small amount of H2O went away between 171 and 306 °C. Nevertheless, ethyl chloride was also detected by TG-MS indicating a decomposition process. Increasing the temperature from 101 °C up to 212 °C resulted in a change in the XRD patterns with gradual formation of amorphous phases. MgOHCl, MgCl2 (aq) probably existed at higher temperatures. The XRD pattern obtained at 500 °C indicated a cubic crystalline material. The pattern of MgO could be fitted well, but the coexistence of other cubic components could also be suggested. The “temperature programmed” XRD behavior of sample 18 was very similar to that of sample 12.
Results of XRD measurements led us to the following conclusions. Mixtures of different phases containing alcohol and H2O existed in all of our salt–alcohol solvate samples. CaCl2 was retrievable from its alcohol solvate by bake out of the alcohol solvate, but irreversible processes appeared in case of MgCl2–alcohol solvates. Upon increasing the temperature up to 500 °C, MgO was obtained with elimination of hydrogen chloride similarly to the MgCl2·6H2O·1,4-C4H8O2 [77] and MgCl2·6H2O system. Although the transformation of the MgCl2 hydrates has long been known [78], clarifying of its mechanism is still in the focus of interest [79].
Possible problems of the applications of salt–alcohol solvates in heat storage systems, based on the results of various techniques used
Our results revealed that the MgCl2–EtOH solvates were instable compound; decomposition of these samples during storage, handling, and analysis was assumed. Raman spectroscopic measurements proved the appearance of ethyl chloride from MgCl2–EtOH without heating. Upon heating, both MeOH and EtOH solvates of MgCl2 were involved in alkyl chloride release, as proven by TG-MS measurements. The alkyl chloride ion current increased slightly from the beginning of the heating and intensified above 140 °C. Although the heating rate can influence on the mechanism of the decomposition, TG-MS measurements by use of high heating rate indicated the formation of alkyl chloride as well as Raman spectroscopy without any heating. On the other hand, XRD measurements showed transformation of MgCl2 in all MgCl2–alcohol solvate samples, which released alkyl chloride.
These observations were in accordance with the literature. Micro-calorimetric analysis has been conducted by Iyimen-Schwarz [51], who has made pioneering scientific contributions to the usability of salt–alcohol solvates based on CaCl2/MgCl2 and MeOH/EtOH for thermal energy storage. Iyimen-Schwarz [51] calculated the energy density from the forward and reverse reaction enthalpy determined in dynamic DSC measurements under vacuum at controlled temperatures and alcohol vapor pressures. Based on observations on both the shape of the MgCl2–MeOH solvate’s measurement curve and the change of the physical appearance, Iyimen-Schwarz suspected the decomposition of the MgCl2–MeOH solvate and the release of CH3Cl with successive cycling, analogous to the reaction of MgCl2 with H2O, affecting the reproducibility of the measurements. According to his work, EtOH can be desorbed easily from the MgCl2–ethanolate system. Iyimen-Schwarz further assumed that in this system the MgCl2 is likely to decompose into C2H5Cl and HCl, due to unreproducible results of the measured dissociation enthalpy. Results of our TG-MS and Raman spectroscopic measurements fully support the conclusions of Iyimen-Schwarz and explain the poor cyclic stability of MgCl2–EtOH solvates [51].
For alkyl chloride formation in our system different pathways could be assumed. It has been reported that gas phase reaction of EtOH and HCl with formation of H2O over ZnCl2/Al2O3 as a catalyst, is a suitable method for ethyl chloride preparation [80]. According to a possible explanation, the starting material (MgCl2) contains MgCl2·zH2O in the presence of traces of H2O. Heating the MgCl2 hydrates results in MgOHxClx with probable formation of hydrochloric acid (HCl). At the same time, reaction of alcohols and HCl can lead to the formation of methyl chloride (CH3Cl), or ethyl chloride (C2H5Cl) via MgCl2 acting as a catalyst. Deliberation of H2O carries on the decomposition of MgCl2. A direct interaction between the ligands (i.e., Cl− and alcohol) in the coordination sphere of the Mg2+ can also be assumed. This idea does not need the H2O to assist in the alkyl chloride formation, but it can lead to the gradual hydrolysis of MgCl2. Although we do not have direct evidence for the first or second pathway, we believe that the direct way is more likely, since ethyl chloride formation appeared even at room temperature, while HCl release from MgCl2·2H2O was reported only at 167 °C [79]. It is worth to note that the final temperature in the cyclic stability test of MgCl2·zEtOH was 180 °C [29].
The results presented in Sect. 3.3–3.5 demonstrate that pure salt–alcohol solvates could not be prepared under technically applicable conditions; the samples contained at least traces of H2O. H2O could be introduced by both the preparation procedure and by the starting materials, despite the use of commercial absolutized solvents and N2 atmosphere for the preparation. The starting salts also contained 1–2% of H2O, which resulted in the presence of salt hydrates besides the mixture of salt–alcohol solvates. The possibility of the formation of salt hydrates implies the possibility of HCl formation during the thermal treatment above 167 °C [81]. In practical applications, an inert atmosphere cannot be maintained during the whole process, so that traces of H2O cannot be avoided. Additionally, the use of high purity grade H2O-free substances might be too expensive at technical scale. The appearance of side reactions and the release of HCl with cycling can contribute to corrosion of the reactor components. The decomposition of MgCl2–alcohol solvates results further in a degradation of the overall performance of the thermal energy storage system. In conclusion, the reaction system MgCl2·R-OH is not suitable for practical implementation, due to its instability and irreversibility. From the energetic point of view, this compound is also not favorable as the measured enthalpies of reaction and associated energy densities are lower than that of CaCl2–alcohol solvates and salt-H2O systems [76].
Conclusions
Reversible chemical reactions are highly efficient in terms of storage volume, storage period, and sensible heat losses to the environment compared to other energy storage technologies. Different CaCl2- and MgCl2–alcohol solvates (EtOH, MeOH) were synthesized and their suitability for heat storage was examined by employing combined thermogravimetric analysis and differential scanning calorimetry (TGA/DSC), spectrometric and spectroscopic analysis (TG-MS, Raman) methods as well as by using X-ray diffraction (XRD). Due to their chemical nature, the CaCl2–EtOH systems exhibited lower energy densities than CaCl2-H2O systems. Decomposition of MgCl2–EtOH solvates accompanied by ethyl chloride formation started already during storage. Upon heating, both MeOH and EtOH solvates of MgCl2 were affected by alkyl chloride release, as proven by TG-MS measurements. Our results fully support the assumptions of Iyimen-Schwarz [51] and explain the poor cycle stability of MgCl2–EtOH solvates reported previously. We also demonstrated that pure salt–alcohol solvates cannot be prepared under technically applicable conditions. Formation of salt hydrates implies the possibility of HCl formation during the thermal treatment. Appearance of side reactions and possible release of HCl with cycling may be conducive to corrosion. Conclusively, MgCl2–alcohol solvate systems are not recommended for heat storage, whereas CaCl2–alcohol systems are suitable demonstrating stable cycle performance. The information on the decomposition pattern and associated changes in the structural integrity of the salt alcoholates obtained in this study are essential for selecting and designing efficient thermochemical energy stores. The fundamental data on the heat content of the parent salt provide the basis for the development of new two-component thermochemical materials with advanced properties.
References
Dincer I, Rosen MA. Thermal energy storage systems and applications. Chichester: Wiley; 2002.
Garg HP, Mullick SC, Bhargava AK. Solar thermal energy storage. Dodrecht: D. Reidel Publishing Company; 1985.
Agyenim F, Hewitt N, Eames P, Smyth M. A review of materials, heat transfer and phase change problem formulation for latent heat thermal energy storage systems (LHTESS). Renew Sustain Energy Rev. 2010;14:615–28.
Feczkó T, Trif L, Horák D. Latent heat storage by silica-coated polymer beads containing organic phase change materials. Sol Energy. 2016;132:405–14.
Kenfack F, Bauer M. Innovative phase change material (PCM) for heat storage for industrial applications. Energy Proc. 2014;46:310–6.
Naumann R, Emons HH. Results of thermal analysis for investigation of salt hydrates as latent heat-storage materials. J Therm Anal Calorim. 1989;35:1009–31.
Naumann R, Schatz O. Thermoanalytical investigation of some alkaline earth hydroxidehydrates on application as latent heat storage materials. J Therm Anal Calorim. 1992;38:665–71.
Sharma A, Tyagi VV, Chen CR, Buddhi D. Review on thermal energy storage with phase change materials and applications. Renew Sustain Energy Rev. 2009;13:318–45.
Zhang N, Yuan Y, Cao X, Du Y, Zhang Z, Gui Y. Latent heat thermal energy storage systems with solid–liquid phase change materials: a review. Adv Eng Mater. 2018;20:1700753. https://doi.org/10.1002/adem.201700753.
Anghel EM, Georgiev A, Petrescu S, Popov R, Constantinescu M. Thermo-physical characterization of some paraffins used as phase change materials for thermal energy storage. J Therm Anal Calorim. 2014;117:557–66.
Kong W, Lei Y, Jiang Y, Lei J. Preparation and thermal performance of polyurethane/PEG as novel form-stable phase change materials for thermal energy storage. J Therm Anal Calorim. 2017;130:1011–9.
Guo L, Yu X, Gao D, Guo Y, Ma C, Deng T. Synthesis and thermal energy storage properties of a calcium-based room temperature phase change material for energy storage. J Therm Anal Calorim. 2018. https://doi.org/10.1007/s10973-018-7610-3.
He Y, Zhang N, Yuan Y, Cao X, Sun L, Song Y. Improvement of supercooling and thermal conductivity of the sodium acetate trihydrate for thermal energy storage with α-Fe2O3 as addictive. J Therm Anal Calorim. 2018;133:859–67.
Cui W, Zhang H, Xia Y, Zou Y, Xiang C, Chu H, et al. Preparation and thermophysical properties of a novel form-stable CaCl2·6H2O/sepiolite composite phase change material for latent heat storage. J Therm Anal Calorim. 2018;131:57–63.
Genc M, Karagoz Genc Z. Microencapsulated myristic acid–fly ash with TiO2 shell as a novel phase change material for building application. J Therm Anal Calorim. 2018;131:2373–80.
Bevers ERT, Oonk HAJ, Haije WG, van Ekeren PJ. Investigation of thermodynamic properties of magnesium chloride amines by HPDSC and TG. J Therm Anal Calorim. 2007;90:923–9.
Jabbari-Hichri A, Bennici S, Auroux A. Water sorption heats on silica-alumina-based composites for interseasonal heat storage. J Therm Anal Calorim. 2014;118:1111–8.
Posern K, Osburg A. Determination of the heat storage performance of thermochemical heat storage materials based on SrCl2 and MgSO4. J Therm Anal Calorim. 2018;131:2769–73.
Posern K, Kaps C. Humidity controlled calorimetric investigation of the hydration of MgSO4 hydrates. J Therm Anal Calorim. 2008;92:905–9.
Lager D, Hohenauer W, Knoll C, Weinberger P, Werner A. Methodology to determine the apparent specific heat capacity of metal hydroxides for thermochemical energy storage. J Therm Anal Calorim. 2018;133:207–15. https://doi.org/10.1007/s10973-017-6883-2.
Bevers ERT, van Ekeren JP, Haije WG, Oonk HAJ. Thermodynamic properties of lithium chloride ammonia complexes for application in a high-lifthigh temperature chemical heat pump. J Therm Anal Calorim. 2006;86:825–32.
Ishitobi H, Uruma K, Takeuchi M, Ryu J, Kato Y. Dehydration and hydration behavior of metal-salt-modified materials for chemical heat pumps. Appl Therm Eng. 2013;50:1639–44.
Donkers PAJ, Pel L, Adan OCG. Experimental studies for the cyclability of salt hydrates for thermochemical heat storage. J Energy Storage. 2016;5:25–32.
Hamdan MA, Rossides SD, Haj Khalil R. Thermal energy storage using thermo-chemical heat pump. Energy Convers Manag. 2013;65:721–4.
Korhammer K, Druske MM, Fopah-Lele A, Rammelberg HU, Wegscheider N, Opel O, Osterland T, Ruck W. Sorption and thermal characterization of composite materials based on chlorides for thermal energy storage. Appl Energy. 2016;162:1462–72.
Posern K, Kaps Ch. Calorimetric studies of thermochemical heat storage materials based on mixtures of MgSO4 and MgCl2. Thermochim Acta. 2010;502:73–6.
Carling RW, Wondolowski AT, Macmillan DC. Enthalpy of formation of CaCl2x2CH3OH and CaCl2x·2C2H5OH by solution calorimetry. J Chem Thermodyn. 1982;14:125–31.
Cho HS, Lee WY. Synthesis of inorganic MgCl2–alcohol adduct via recrystallization method and its application in supported organometallic catalysts for the polymerization of ethylene with 1-hexene. J Mol Catal A-Chem. 2003;191:155–65.
Korhammer K, Apel C, Osterland T, Ruck WKL. Reaction of calcium chloride and magnesium chloride and their mixed salts with ethanol for thermal energy storage. Energy Proc. 2016;91:161–71.
Muroyama AP, Schrader AJ, Loutzenhiser PG. Solar electricity via an Air Brayton cycle with an integrated two-step thermochemical cycle for heat storage based on Co3O4/CoO redox reactions II: kinetic analyses. Sol Energy. 2015;122:409–18.
Olivares R, Edwards W. LiNO3–NaNO3–KNO3 salt for thermal energy storage: thermal stability evaluation in different atmospheres. Thermochim Acta. 2013;560:34–42.
Kousksou T, Jamil ĆA, Gibout ĆS, Zeraouli YĆ. Thermal analysis of phase change emulsion. J Therm Anal Calorim. 2009;96:841–52.
Anghel EM, Georgiev A, Petrescu S, Popov R, Constantinescu M. Thermo-physical characterization of some paraffins used as phase change materials for thermal energy storage. J Therm Anal Calorim. 2014;117:557–66.
Wong B, Brown L, Buckhingham R, Sweet W, Russ B, Gorensek M. Sulfur dioxide disproportionation for sulfur based thermochemical energy storage. Sol Energy. 2015;118:134–44.
van Essen VM, Zondag HA, Gores JC, Bleijendaal LPJ, Bakker M, Schuitema R, van Helden WGJ, He Z, Rindt CCM. Characterization of MgSO4 hydrate for thermochemical seasonal heat storage. J Sol Energy Eng. 2009;131:041014-1–7.
van Essen VM, Bleijendaal LPJ, Kikkert BWJ, Zondag HA, Bakker M, Bach PW. Development of a compact heat storage system based on salt hydrates. In: Proceedings of the EUROSUN; 2010.
Zondag H, van Essen M, Bleijendaal L, Cot J, Schuitema R, Planje W, Epema T, Oversloot H. Comparison of reactor concepts for thermochemical storage of solar heat. In: Third International Renewable Energy Storage Conference, IRES; 2008:24–5.
Aristov YI, Gordeeva LG, Pankratiev YD, Plyasova LM, Bikova IV, Freni A, Restuccia G. Sorption equilibrium of methanol on new composite sorbents “CaCl2/silica gel”. Adsorption. 2007;13:121–7.
Gillier-Pandraud H, Philoche-Levisalles M. Structure cristalline du compose CaCl2 CH3OH. C R Acad Sci Ser C. 1971;273:949–51.
Hirata Y, Fujioka K. Thermophysical properties and heat transfer characteristics of CaCl2 heat pump reactor associated with structural change of reactive salts. In: V Minsk international seminar ‘‘Heat pipes, heat pumps, refrigerators”; 2003.
Lai H, Li C. Application of periodic reversal flow reactors to chemical heat pump systems based on solid/vapor non-catalytic reaction. Chem Eng Sci. 1996;51:2951–7.
Offenhartz PO’D, Brown FC, Mar RW, Carling RW. A heat pump and thermal storage system for solar heating and cooling based on the reaction of calcium chloride and methanol vapor. J Sol Energy Eng. 1980;102:59–65.
Gmelin L. Handbuch der anorganischen chemie. Weinheim: Verlag Chemie; 1966.
Bonnell DGR, Jones WJXLII. The dissociation pressures of alcoholates. Part I. J Chem Soc (Resumed). 1926;129:321–8.
Menschutkin BN. Über einige Kristallalkoholate. Zeitschrift für anorganische und allgemeine Chemie. 1907;52:9–24.
Brusset H, Gillier-Pandraud H, Philoche-Levisalles M. Crystallographic data on compound CaCl2. C R Acad Sci Ser C. 1970;271:579.
Halut-Desportes S, Husson E. Spectres d’absorption infrarouge et calcul de champ de force de complexes de coordination: halogénures de magnésium et de calcium avec le méthanol ou l’éthanol. Spectrochim Acta A Mol Spectr. 1985;41:661–73.
Kane R. Beiträge zur Geschichte des Holzgeistes und seiner Verbindungen. Eur J Org Chem. 1836;19:164–83.
Halut-Desportes S, Philoche-Levisalles M. Structures comparées des solvates de formule MBr2. 6CH3OH. Acta Crystallogr B-Struct Crystallogr Cryst Chem. 1978;34:432–5.
Mar RW, Carling RW. The calcium chloride—ethanol system. Thermochim Acta. 1981;45:213–7.
Iyimen-Schwarz, Z. Energiespeicherung durch chemische Reaktionen. Doctoral dissertation; 1984.
Heindl JB. Über krystallinische Verbindungen von Chlorcalcium mit Alkoholen. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften. 1881;2:200–11.
Parker VB, Wagman DD, Evans WH. NBS Technical Note 270-6. Selected values of thermodynamic properties. Washington: US Government Printing Office; 1971.
von Chodnew A. Beiträge zur Kenntniss der Alkoholate und der salpetersauren Magnesia. Eur J Org Chem. 1849;71:241–65.
Graham T. XLVII. An account of the formation of alcoates, definite compounds of salts and alcohol analogous to the hydrates. Philos Mag Ser. 1828;2(4):265–72.
Multani RK. Action of magnesium chloride on alcohols. Curr Sci. 1964;33:430.
Olmer LJ, Quinet MLB. Soc Chim Fr. 1934;1:1579–81.
Choi JH, Chung JS, Shin HW, Song IK, Lee WY. The effect of alcohol treatment in the preparation of MgCl2 support by a recrystallization method on the catalytic activity and isotactic index for propylene polymerization. Eur Polym J. 1996;32:405–10.
Quinet MLB. Soc Chim Fr. 1936;3:1829.
Emons HH, Pollmer K. Studies on systems of salts and mixed solvents. XXXI. On the solubility and solvation behaviour of magnesium chloride in mixed aqueous organic solvents. Zeitschrift für anorganische und allgemeine Chemie. 1985;521:224–30.
Gnanakumar ES, Gowda RR, Kunjir S, Ajithkumar TG, Rajamohanan PR, Chakraborty D, Gopinath CS. MgCl2·6CH3OH: a simple molecular adduct and its influence as a porous support for olefin polymerization. ACS Catal. 2013;3:303–11.
Kabisch G, Bader I, Emons HH, Pollmer K. A Raman spectroscopic investigation of the structure of magnesium salt solutions in methanol and methanol-water mixtures. J Mol Liq. 1983;26:139–57.
Lloyd E, Brown CB, Bonnell DGR, Jones WJ. XC.—Equilibrium between alcohols and salts. Part II. J. Chem. Soc. (Resumed). 1928;658–66. https://doi.org/10.1039/JR9280000658
Radnai T, Kalman E, Pollmer K. X-ray diffraction study of MgCl2 in methanol. Z Naturforsch Pt A. 1984;39:464–70.
Simon SE. Über die Verbindungen des Chlorlithiums und des Chlormagnesiums mit Alkoholen. Adv Synth Catal. 1879;20:371–7.
Turova NY, Turevskaya EP. Alkoxymagnesium halides. J Organomet Chem. 1972;42:9–17.
Nasirzadeh K, Zimin D, Neueder R, Kunz W. Vapor-pressure measurements of liquid solutions at different temperatures: apparatus for use over an extended temperature range and some new data. J Chem Eng Data. 2004;49:607–12.
Tewell CR, Malizia F, Ager JW, Somorjai GA. An ultraviolet–Raman spectroscopic investigation of magnesium chloride–ethanol solids with a 0.47 to 6 molar ratio of C2H5OH to MgCl2. J Phys Chem B. 2002;106:2946–9.
Chadwick JC, Severn JR. Single-site catalyst immobilization using magnesium chloride supports. Kinet Catal. 2006;47:186–91.
Bart JCJ, Roovers W. Magnesium chloride-ethanol adducts. J Mater Sci. 1995;30:2809–20.
Di Noto V, Zannetti R, Vivani M, Marega C, Marigo A, Bresadola S. MgCl2-supported Ziegler–Natta catalysts: a structural investigation by X-ray diffraction and Fourier-transform IR spectroscopy on the chemical activation process through MgCl2-ethanol adducts. Macromol Chem Phys. 1992;193:1653–63.
Malizia F, Fait A, Cruciani G. Crystal structures of Ziegler–Natta catalyst supports. Chem Eur J. 2011;17:13892–7.
Jiang X, Tian X, Fan Z. Crystal structure of ball-milled mixture of sodium chloride and magnesium chloride–ethanol adduct. Mater Res Bull. 2008;43:343–52.
Taveira Magalhaes DN, Do Coutto Filho O, Coutinho FMB. Ziegler-Natta catalyst for ethylene and propylene polymerization supported on adducts of magnesium chloride with methyl and ethyl alcohols. Eur Polym J. 1991;27:827–30.
Thushara KS, Gnanakumar ES, Mathew R, Ajithkumar TG, Rajamohanan PR, Bhaduri S, Gopinath CS. MgCl2• 4((CH3)2CHCH2OH): a new molecular adduct for the preparation of TiClx/MgCl2 catalyst for olefin polymerization. Dalton Trans. 2012;41:11311–8.
N’Tsoukpoe KE, Schmidt T, Rammelberg HU, Watts BA, Ruck WK. A systematic multi-step screening of numerous salt hydrates for low temperature thermochemical energy storage. Appl Energy. 2014;124:1–16.
Jin M, Sun Y, Li P, Yu J, Ulrich J. The thermal decomposition study of MgCl2·6H2O·1,4-C4H8O2. Chem Eng Res Des. 2015;104:256–63.
Galwey AK, Laverty GM. The thermal decomposition of magnesium chloride dihydrate. Thermochim Acta. 1989;138:115–27.
Huang Q, Lu G, Wang J, Yu J. Mechanism and kinetics of thermal decomposition of MgCl2x6H2O. Metall Mater Trans B. 2010;41:1059–66.
Bukhanko N, Samikannu A, Larsson W, Shchukarev A, Leino AR, Kordás K, Warna J, Mikkola JP. Continuous gas-phase synthesis of ethyl chloride from ethyl alcohol and hydrochloric acid over Al2O3 based catalysts: the “green” route. ACS Sustain Chem Eng. 2013;1:883–93.
Huang Q, Lu G, Wang J, Yu J. Thermal decomposition mechanisms of MgCl2·6H2O and MgCl2·H2O. J Anal Appl Pyrol. 2011;91:159–64.
Wagman DD, Evans WH, Parker VB, Schumm RH, Halow I. The NBS tables of chemical thermodynamic properties. Selected values for inorganic and C1 and C2 organic substances in SI units. National Standard Reference Data System, 1982.
Acknowledgements
Open access funding provided by MTA Research Centre for Natural Sciences (MTA TTK). Project no. TÉT_12_DE-1-2013-0003 has been implemented with the support provided by the National Research, Development and Innovation Fund of Hungary, financed under the TÉT_12_DE funding scheme. This study received funding from the German Federal Ministry of Education and Research (BMBF) within the framework of a bilateral research collaborative project between the Leuphana University of Lüneburg and the Hungarian Academy of Sciences under grant agreement number 01DS14029. The authors thank Christina Apel for the preparation of salt–alcohol solvate samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix 1: Calculations
The alcohol/salt molar ratios (levels of alcoholation) were derived from experimental data obtained by TGA. The percentage mass loss X is defined as the mass of alcohol mR-OH desorbed per unit mass of salt alcoholate mMX ZR-OH:
The level of alcoholation z is defined as the ratio of the number of alcohol molecules evolved during the endothermic dissociation reaction nR-OH to the amount of anhydrous salt nMX and was calculated from the following equation:
wherein mMX and mR-OH are the masses of the anhydrous salt and the alcohol desorbed, respectively.
The principle of thermochemical heat storage using reversible gas–solid reactions is based on the conversion of thermal energy into chemical energy required to break the chemical bonds of the reactants. At constant pressure, the amount of heat energy that must be supplied to induce the decomposition reaction equals the endothermic heat of reaction, also designated enthalpy of reaction. An infinitesimal change in the temperature results in a change of the enthalpy by ΔCpdT. The enthalpy of reaction ΔrH under non-standard condition, in case of a reaction temperature T1 different from the standard state temperature T0 can be estimated from the standard reaction enthalpies and heat capacities of the reactants using Kirchhoff’s law. The heat energy ΔQ is then expressed by:
where ΔrH0(T0) is the standard enthalpy of reaction at standard state conditions T0 = 298.15 K and p0 = 1 bar, Cp is the constant-pressure heat capacity and ΔTrH is the enthalpy of transformation.
The enthalpy of reaction was obtained from DSC measurement by peak area integration. The energy that is liberated or absorbed as heat during a chemical reaction as a result of a temperature difference ΔT can be quantitatively determined by heat flux DSC. This technique measures the thermally induced heat flux transferred between the sample and an inert reference that are connected by a low-resistance heat flow path. Thermocouples below the symmetrically positioned sample crucible and empty reference crucible detect and compare the temperature of the specimen to the temperature of the reference as a function of time under same conditions. The heat flux between sample and reference is proportional to the temperature difference:
wherein E(T) and DSC are the calorimetric sensitivity and the measured DSC signal. Integration of the peak area under the baseline-subtracted DSC signal over time yields ΔrH0:
According to Hess Law, the value of the standard reaction enthalpy of the forward and reverse reaction must be equal and the same applies for the standard enthalpy of association and standard enthalpy of dissociation. The standard enthalpy of dissociation is the inverse of the standard enthalpy of formation. In general, the standard enthalpy of reaction is calculated from the difference of the standard enthalpy of formation of the products and the standard enthalpy of formation of the reactants:
Solving Eq. (8) for \(\Delta_{\text{f}} H_{\text{products}}^{0}\) gives the standard enthalpy of formation and hence the standard enthalpy of dissociation.
Different mathematical approaches are used to determine the specific energy storage density. Two types of energy densities are known: gravimetric and volumetric energy density. The gravimetric energy density Em is defined as the capacity of heat energy stored at a defined temperature and pressure per unit mass of storage material and can be calculated from the ratio of the standard reaction enthalpy to the molar mass of the reactant MMX zR-OH:
whereas the volumetric energy density EV is related to the volume of storage material and is described as:
The storage material volume is derived from the material’s mass and bulk density ρ. The higher the level of alcoholation, the lower is the density. The volumetric energy density is an important key energy storage metric for designing and operating storage systems. It is also preferred for performance comparison studies. Space can be a limiting factor for many practical applications. There are no data available for the mass and bulk densities of the salt–alcohol systems studied and hence only the gravimetric energy density could be calculated.
Appendix 2: Data processing—example of a TGA/DSC curve
Figure 16 displays the DSC measurement curve and corresponding TGA signal normalized to the sample mass for sample 6. To determine the heat content of the sample and the associated enthalpy of reaction, the area under the DSC peak was integrated. Similar curves were recorded for all other samples.

Example of a recorded TGA and DSC curve. The TGA curve or change in mass (%) and DSC curve or heat flow (mW) are given as a function of time (min) and temperature (°C). The sample was heated from 30 to 180 °C at a heating rate of 3 K min−1. Since the signs of the thermal processes were inverted due to software issues, the signed number of the underlying endothermic reaction is negative
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Korhammer, K., Mihály, J., Bálint, S. et al. Reversible formation of alcohol solvates and their potential use for heat storage. J Therm Anal Calorim 138, 11–33 (2019). https://doi.org/10.1007/s10973-019-08090-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08090-2