
Abstract
Compounds of poly(butylene terephthalate) (PBT) and zinc oxide (ZnO) with filler content between 1 and 10% were prepared in a laboratory internal mixer. The processing parameters did not damage PBT thermal stability or its molecular weight as evidenced by torque rheology. The melt crystallization of PBT/ZnO compounds was investigated by differential scanning calorimetry and their morphology by scanning electron microscopy and optical microscopy. From morphological analyses, ZnO particles are well dispersed in PBT matrix, which crystallizes in typical spherulites. The melt crystallization temperatures and maximum melt crystallization rates are almost unaffected by the filler. Equally the overall crystallinity did not show any dependence on the filler content or the cooling rate. Mo’s model was found to be suitable for a description of the melt crystallization kinetics, while Ozawa model turned out to be inadequate.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(butylene terephthalate) (PBT) is a semicrystalline aromatic polyester, well known as a prime material in the industrial polymer field which needs engineering thermoplastics with excellent dimensional stability, high strength, chemical resistance and fast crystallization rate. To extend even more its applications, compounding with nanofillers like organoclays and carbon nanofibers [1], montmorillonite [2], calcium carbonate (CaCO3) [3], nano SiO2 [4], talc and wollastonite [5] have been reported in the literature. In most cases, addition of nanofillers improves the crystallization rate and lowers the onset and peak crystallization temperatures of PBT; for highly loaded compounds, slower crystallization rates and growth processes have been verified. These effects are dependent on the amount of filling used and on the cooling rate applied, with more pronounced changes noticed at higher cooling rates. To the best of our knowledge, crystallization of PBT/ZnO compounds has not been reported in the literature. ZnO is an inorganic and inert compound widely used as an additive in numerous materials and products. In the field of polymer science, ZnO is known to present high refractive index, high thermal conductivity, binding, antibacterial action (antiseptic) and UV protection, transferring properties to its polymer compounds [6,7,8,9,10].
To obtain plastic products that meet stringent requirements, it is important to know relationships between processing conditions, crystallization behavior, structural and morphological development and its final properties. Crystallization is the main process from which structure and morphology develop. Understanding the crystallization kinetics of polymer nanocomposites is of practical importance. Polymer processing generally involves nonisothermal crystallization conditions; therefore, nonisothermal crystallization study is crucial from a scientific prospective.
In the present contribution, the nonisothermal melt crystallization of virgin PBT and PBT/ZnO nanocomposites was investigated using differential scanning calorimetry (DSC), and the resulting morphologies were analyzed by optical microscopy (OM) and scanning electron microscopy (SEM). Additionally the effect of processing parameters on the thermal integrity as well as on the molecular weight of PBT was studied by torque rheometry.
The main objective of this work is to understand the effect of ZnO filler content and DSC cooling rates on the PBT crystallization. The presented kinetic modeling is based on the Ozawa [11] and Mo [12, 13] macrokinetic models.
Experimental
Materials
The PBT used was 195 Valox, gently supplied by Sabic company (Bergen op zoom Netherlands), with density 1.31 g cm−3 and melting temperature T m = 225 °C.
Zinc oxide (ZnO) was purchased from Acros Organics (Brazil) (MDL Number MFCD00011300, 99.5% purity). It has a specific area of 28 m2 g−1. In this work, ZnO was incorporated into the PBT matrix without using any dispersion agent.
Compounding
PBT compounds containing 1, 5 and 10% per mass ZnO were prepared in a Haake Rheomix 600 laboratory internal mixer fitted with high intensity rotors (type: rollers). Table 1 summarizes the range of experimental parameters such as ZnO content, chamber temperature, nominal rotor speed and processing time.
Scanning electron microscopy
Scanning electron microscopy images were acquired with a LEO 1430 unit, from Zeiss. The polymer samples were fractured in liquid nitrogen to avoid plastic deformation and coated with a carbon layer in order to avoid the accumulation of charges.
Optical microscopy
Optical microscopy images were obtained from thin slices cut from PBT compounds using a Hinox model KH 1300 series H05008. The selected samples were melted on glass slides in a hot stage IKA Model C-Mag HS4 setup at 240 °C, and images were acquired during and after cooling.
DSC analyses
Nonisothermal crystallization kinetics was performed using a TA Instruments DSC Q20 differential scanning calorimeter, under a nitrogen flow of 50 mL min−1. The instrument was calibrated with high-purity indium standard. For melt crystallization, the samples were held at 250 °C for 3 min in order to remove small nuclei that might act as seeding crystals. Then, the samples were cooled down under different rates of 5, 10, 20 and 30 °C min−1.
For each crystallization peak, the starting and end points of departure from the underlying baseline were visually established in a plot of energy flow (J) versus time (t). The fractional crystallinity x was computed as a function of time by integration:
where J 0 is the virtual baseline during the event (a straight line in the present case), and E 0 is the total latent heat of the phase change:
and t 1 and t 2 are the initial and final times. The crystallization rate c is:
from which the peak (maximum) and average crystallization rates may be computed.
The specific latent heat of crystallization or melting (or enthalpy, because the phase change occurs at constant pressure) is computed from E 0, the polymer fraction w P and the sample mass m S:
The mass crystallinity change ∆X c during the event is estimated, taking into account the heat of fusion of PBT 100% crystalline:
A value of \(H_{\text{m}}^{0} = 140\) J g−1 at the equilibrium melting temperature \(T_{\text{m}}^{0}\) = 249 °C was previously reported in the literature [14].
Results and discussion
Torque rheometry analysis
In this work, the objective of torque rheometry analysis is to gather information regarding polymer degradation during mixing. Considering the last processing stage, the torque Z is related to melt viscosity η:
which is proportional to a high power of the average molar mass M of the polymer. For any molten polymer characterized by the pseudoplastic index n, the following equation holds approximately:
Constants k 1 and k 2 are intrinsic processing parameters, depending on mixer geometry, processing conditions, polymer and temperature [15].
The observed viscosity or torque during the final stage of melt mixing depends on two parameters: temperature and molar mass; this means, in order to attribute a decrease in viscosity to polymer degradation, the effect of temperature on viscosity has to be eliminated. This was done using a temperature-adjusted torque Z*:
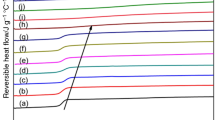
Here, T* is an arbitrary reference temperature and β is called coefficient of viscosity. The parameter β was determined experimentally considering a series of processing experiments termination at different temperatures T end. It was calculated from a linear fit of ln Z taken at T end versus T end − T* (see Fig. 1). T* is ideally the mean of the temperature range considered for T end (in this work 240 °C).

Determination of β via linear fit of ln Z versus T end − T* (β = 0.027 ± 0.002 °C−1)
Next, for a 2-min time interval (last processing stage between 8 and 10 min), the rate of change of the temperature-corrected torque Z* is determined, and the relative rate of change of adjusted torque R Z is defined as:
where \(\overline{{Z^{*} }}\) is the mean value of Z* over the selected time interval. Similarly, for the relative rate of change of the mean molar mass \(\overline{M}\) holds approximately:
One hundred times R M represents the percent variation of the mean molar mass per unit processing time.
The pseudoplastic index n can be expressed my means of the shear stress τ:
where κ is the flow consistency index and \(\frac{\partial u}{\partial y}\) is the velocity gradient (perpendicular to the plane of shear). As shear stress and velocity gradient are proportional to torque and shaft rotation velocity (turns per unit time, N), n can be determined via linear regression of ln Z* versus ln N, as shown in Fig. 2.

Determination of n via linear fit of ln Z* versus ln N. N is the number of turns per unit time (n = 0.948 ± 0.015)
The utilization of the adjusted torque Z* is justified as follows: The equation is valid only for steady one-dimensional shear flow, u = u x = u x(y), u y = u z = 0 and τ = τ xy = τ yx in which case the equation is better expressed in terms of an ordinary derivative. Moreover, the equation is valid only if the fluid strictly follows a power law (which is more restrictive requirement than being a pseudoplastic fluid). Molten polymers follow a power law only within a range deformation rates. Even if n were independent of temperature, Z would be proportional to N n only at constant temperature. For a power-law melt with all other variables constant, the following equation holds approximately:
The problem is handled by defining an “adjusted” torque Z*:
which is, in fact, proportional to N n and independent of temperature (for a stable melt, and assuming all the restrictive conditions mentioned earlier). More examples how to apply this methodology can be found in references [16,17,18,19,20].
Table 2 presents the numerical values for R Z and R M, which are close to zero. With exemption of the composition PBT/5% ZnO, these values exhibit positive sign which is an unphysical increase in molecular weight. All these values are in the order of the measurement accuracy, indicating that no polymer degradation can be observed during melt mixing.
SEM analysis
Figure 3 presents SEM images of neat PBT and PBT + 10% ZnO; it can be confirmed that ZnO particles are quite homogeneously distributed in the PBT matrix. They are visible as small white spots.

Scanning electron micrograph of a neat PBT and b PBT/10% ZnO, as obtained after mixing
DSC measurements
The DSC reveals that both neat PBT and all investigated PBT–ZnO compounds crystallize completely during cooling, i.e., there is no cold crystallization event visible from the scans. DSC scans for neat PBT as well as for PBT/ZnO compounds are given in the Supplemental Figure S1.
Figure 4 shows the effect of ZnO content on PBT, and its compounds melt crystallized at 5 °C min−1 and at 30 °C min−1, respectively. The ZnO content added to PBT in this work ranged from 1 to 10% per mass, so that it may be viewed as an additive at the lowest content and as a filler at higher concentrations. As can be seen, ZnO has very little influence on the sigmoids, behaving like an additive apparently inert to PBT crystallization.

Relative crystallinity versus temperature for melt crystallization at a 5 °C min−1, and b 30 °C min−1. Compositions indicated
By means of integration of the crystallization peak, the total heat of phase transformation is obtained for every event. Knowledge of the equilibrium melting enthalpy of a 100% crystallized PBT sample allows the calculation of the degree of crystallinity. From the evolution of crystallinity with time, the crystallization half times and maximum rates were calculated. The temperature of crystallization peak maximum was directly extracted from the DSC data.
Figure 5 summarizes these data; as can be seen, the crystallization half times and maximum crystallization rates are almost unaffected by the filler. The melt crystallization peak temperatures decrease linearly with an increase in the cooling rate and are equally independent of the filler content. The degree of crystallinity neither shows a consistent dependence on the cooling rate nor on the filler content; for the tested samples, it is within the range of 14–20%.

Melt crystallization parameters for all compositions: a crystallization half time τ 1/2, b crystallization peak temperature T c, c maximum crystallization rate c max and d obtained total crystallinity ΔX c
The results presented in this work are quite interesting, ZnO behaved as an inert filler not degrading PBT during processing or changing its crystallization. From literature, usually the filler acts as nucleating agent, shifting the melt crystallization temperature to higher values favoring the melt crystallization; see for instance a study of PBT compounded with four different nanofillers [21].
Chen et al. [22] found that high loading of PBT with carbon fibers up to 20% per mass increased the latent heat of crystallization, which means the filler increased the final degree of crystallinity. (These authors did got give numerical values for the final degree of crystallinity.) Regarding results shown in the present work, this suggests that ZnO interacts less than carbon fibers with the polymer matrix.
Finally, a study on PBT/halloysite nanotube composites [23] is fully consistent with our results; these authors report a final degree of crystallinity between 25 and 30% for neat PBT, which remains quit unaffected by halloysite loading up to 10% per mass.
Wu et al. [24] reported crystallization half times of 1.20–0.4 min for cooling rates of 5 and 20 °C min−1, respectively. These values are almost equal to those reported in the present study.
Optical microscopy images
Optical microscopy (OM) images of neat PBT and PBT/ZnO compounds were taken immediately after cooling of a molten sample in ambient air to room temperature.
ZnO increases the spherulite size (Fig. 6). However, in both cases, significant residues of uncrystallized polymer are visible.

Optical microscopy images of neat PBT (a) and PBT +10% ZnO (b) after cooling
This result is unexpected although ZnO addition has changed neither the processing parameters (Table 2) nor the crystallization behavior as presented in Fig. 5a–d; it modifies the spherulite size. Comparing with others works, for instance, when loading PBT with calcium carbonate nanoparticles, an increase in filler content leads to a higher number of smaller spherulites per unit area [25]. Consequently [25] suggests that calcium carbonate particles act as nucleation sites in the polymer matrix. However, when compounding PBT with talc particles [26], melt crystallization leads to increase in spherulite size when compared to neat PBT, pointing to a decreased nucleation efficiency of the filler.
Quantitative description of melt crystallization
Mo’s equation [12, 13] relates the time necessary to attain a certain value of relative crystallinity x to a constant heating or cooling rate φ:
where F = F(x) and α = α(x) are the Mo parameters, which can be obtained via linear regression:
As an example, the Mo plot (ln Φ vs. ln t) is shown in Fig. 7 for the compound PBT/5% ZnO (see supplemental materials for additional Mo plots). The data points can be fitted in a reasonable way by straight lines. The variation of the Mo coefficient α and ln F with respect to the relative crystallinity x is plotted in Fig. 8; for numerical values see Tables TS1–TS4 in the supplemental material.

Mo plot ln (φ) versus ln (t) for PBT + 5% ZnO compound considering relative crystallinities from 10 to 90%

Dependence of the Mo exponent α (a) and ln F (b) on the relative crystallinity x for neat PBT and PBT/ZnO compounds
Notably the Mo exponent alpha is strongly dependent on the filler content and varies very little with crystallinity. The implication for practical kinetic modeling is that for every composition, the Mo exponent could be approximated as a constant, and this for instance could be the mean value.
The parameter ln F could be expressed as a linear function of the relative crystallinity. ln F seems to be independent of the filler content; however, it can be distinguished between neat PBT and any PBT–ZnO compound. In any case, a determination of the Mo exponent is mandatory for every filler loading level.
The kinetics of neat PBT described by Mo model has already been reported in the literature [24]. Similarly to our results, the Mo exponent was found almost constant (1.12–1.13) within the relative crystallinity range from 20 to 80%; in this work Mo’s exponent varied between 1.4 and 1.5 for neat PBT. Equally the parameter ln F did not vary.
Ozawa model defines a dependency of the relative crystallinity x at a constant temperature with the cooling rate Φ:
where κ(T) is a parameter of the nonisothermal crystallization and m is the Ozawa exponent. This equation can be rewritten as
which makes it possible to extract m and Φ via linear regression.
As an example, Fig. 9 shows the Ozawa plot for PBT/5% ZnO at three indicated temperatures. As can be seen, a linear correlation of the data points is poorly fulfilled, indicating that the Ozawa method is inadequate for the description of the melt crystallization kinetics. This is in agreement with the publications by Deshmukh et al. [25] and Zhang et al. [27], which investigated the crystallization kinetics of neat PBT as well as PBT filled with nano-calcium carbonate and nano-SiO2, respectively. Both studies found Ozawa model unsuitable for the description of nonisothermal crystallization kinetics. However, two other studies demonstrated a limited applicability of Ozawa method to PBT crystallization. According to Wu et al. [24], the method failed to describe the neat PBT kinetics, but turned out to be useful for the description of PBT/montmorillonite compounds in a small temperature interval from 186 to 200 °C. Another study by Oburoglu et al. [23] dealing with PBT filled with halloysite nanotubes found Ozawa’s model is adequate for neat PBT in a narrow temperature range from 190 to 202 °C; however, the model failed when filler was added.

Ozawa plot \(\ln \left[ { - \ln \left( {1 - x} \right)} \right]\) versus \(\ln \;\varPhi\) at the indicated temperatures for the composition PBT/5% ZnO
In summary, all these results point to the fact that ZnO does not interact significantly with PBT; it might transfer only additional properties to the final product, such as antibacterial properties and white color, while the processing parameters and crystallization properties remain basically unaltered.
Conclusions
The processing parameters did not damage PBT thermal stability or its molecular weight; PBT/ZnO compounds may be processed as the same way as the neat PBT. The addition of ZnO has no significant effect on the melt crystallization temperatures and maximum rates. The total crystallinity as obtained after melt crystallization does not exhibit any dependence on the filler content or the cooling rate. Mo’s model can be used for modeling the melt crystallization behavior.
References
Al-Mulla A, Mathew J, Yeh S-K, Gupta R. Nonisothermal crystallization kinetics of PBT nanocomposites. Compos A. 2008;39:204–17.
Acierno D, Scarfato P, Amendola E, Nocerino G, Costa G. Preparation and characterization of PBT nanocomposites compounded with different montmorillonites. Polym Eng Sci. 2004;44:1012–8.
Deshmukh GS, Peshwe DR, Pathak SU, Ekhe JD. Nonisothermal crystallization kinetics and melting behavior of poly (butylene terephthalate) and calcium carbonate nanocomposites. Thermochim Acta. 2015;606:66–76.
Che J, Luan B, Yang X, Lu L, Wang X. Graft polymerization onto nano-sized SiO2 surface and its application to the modification of PBT. Mater Lett. 2005;59:1603–9.
Deshmukh GS, Peshwea DR, Pathaka SU, Ekheba JD. Nonisothermal crystallization kinetics and melting behavior of poly(butylene terephthalate) (PBT) composites based on different types of functional fillers. Thermochim Acta. 2014;581:41–53.
Díez-Pascual M, Díez-Vicente AL. Poly(3-hydroxybutyrate)/ZnO bionano-composites with improved mechanical, barrier and antibacterial properties. Int J Mol Sci. 2014;17:10950–73.
Yu W, Lan C, Wang S, Fang P, Sun Y. Influence of zinc oxide nanoparticles on the crystallization behavior of electrospun poly(3-hydroxybutyrate-co-3-hydroxyvalerate) nanofibers. Polymer. 2010;51:2403–9.
Hernandezbattez A, Gonzalez R, Viesca J, Fernandez J, Diazfernandez J, MacHado A, Chou R, Riba J. CuO, ZrO2 and ZnO nanoparticles as antiwear additive in oil lubricants. Wear. 2008;265:422.
Padmavathy N, Vijayaraghavan R. Enhanced bioactivity of ZnO nanoparticles—an antimicrobial study. Sci Technol Adv Mater. 2008;9:035004.
Chen P, Zhou H, Liu W, Zhang M, Du Z, Wang X. The synergistic effect of zinc oxide and phenylphosphonic acid zinc salt on the crystallization behavior of poly (lactic acid). Polym Degrad Stab. 2015;122:25–35.
Ozawa T. Kinetics of non-isothermal crystallization. Polymer. 1971;12:150–8.
Liu T, Mo Z, Wang S, Zhang H. Nonisothermal melt and cold crystallization kinetics of poly(aryl ether ether ketone ketone). Polym Eng Sci. 1997;37:568–75.
Liu T, Mo Z, Wang S, Zhang H. Nonisothermal crystallization behavior of a novel poly(aryl ether ketone): PEDEKmK. J Appl Polym Sci. 1998;67:815–21.
Righetti MC, Di Lorenzo ML, Angiuli M, Tombari E, La Pietra P. Poly(butylene terephthalate)/poly(e-caprolactone) blends: Influence of PCL molecular mass on PBT melting and crystallization behavior. Eur Polym J. 2007;43:4726–38.
Alves TS, Neto JES, Silva SML, Carvalho LH, Canedo EL. Process simulation of laboratory internal mixers. Polym Test. 2016;50:94–100.
Costa ARM, Almeida TG, Silva SML, Carvalho LH, Canedo EL. Chain extension in poly(butylene-adipate-terephthalate). Inline analysis in a laboratory internal mixer. Polym Test. 2015;42:115–21.
Duarte IS, Tavares AA, Lima PS, Andrade DLACS, Carvalho LH, Canedo EL, Silva SML. Chain extension of virgin and recycled poly(ethylene terephthalate): effect of processing conditions and reprocessing. Polym Degrad Stab. 2016;124:26–34.
Tavares AA, Silva DFA, Lima PS, Andrade DLACS, Silva SML, Canedo EL. Chain extension of virgin and recycled polyethylene terephthalate. Polym Test. 2016;50:26–32.
Almeida TG, Neto JES, Costa ARM, Silva AS, Carvalho LH, Canedo EL. Degradation during processing in poly(butylene adipate-co-terephthalate)/vegetable fiber compounds estimated by torque rheometry. Polym Test. 2016;55:204–11.
Marinho VAD, Pereira CAB, Vitorino MBC, Silva AS, Carvalho LH, Canedo EL. Degradation and recovery in poly(butylene adipate-co-terephthalate)/thermoplastic starch blends. Polym Test. 2017;58:166–72.
Yin H, Dittrich B, Farooq M, Kerling S, Wartig K-A, Hofmann D, Huth C, Okolieocha C, Altstädt V, Schönhals A, Schartel B. Carbon-based nanofillers/poly(butylene terephthalate): thermal, dielectric, electrical and rheological properties. J Polym Res. 2015;22:140.
Chen Y, Wang X, Wu D. Recycled carbon fiber reinforced poly(butylene terephthalate) thermoplastic composites: fabrication, crystallization behaviors and performance evaluation. Polym Adv Technol. 2013;24:364–75.
Oburoğlu N, Ercan N, Durmus A, Kaşgöz A. Effects of halloysite nanotube on the mechanical properties and nonisothermal crystallization kinetics of poly(butylene terephthalate) (PBT). J Macromol Sci B. 2012;51:860–79.
Wu D, Zhou C, Fan X, Mao D, Bian Z. Nonisothermal crystallization kinetics of poly(butylene terephthalate)/montmorillonite nanocomposites. J Appl Polym Sci. 2006;99:3257–65.
Deshmukh GS, Peshwe DR, Pathak SU, Ekhe JD. Nonisothermal crystallization kinetics and melting behavior of poly(butylene terephthalate) and calcium carbonate nanocomposites. Thermochim Acta. 2015;606:66–76.
Deshmukh GS, Peshwe DR, Pathak SU, Ekhe JD. Nonisothermal crystallization kinetics and melting behavior of poly(butylene terephthalate) (PBT) composites based on different types of functional fillers. Thermochim Acta. 2014;581:41–53.
Zhang L, Hong Y, Zhang T, Li C. Nonisothermal crystallization behaviors of poly(butylene terephthalate) nucleated with elastomer-modified nano-SiO2 a commercial nucleating agent (P250), and talc. J Macromol Sci, Phys. 2010;49:514–27.
Acknowledgements
The authors would like to thank to José William de Lima Souza (CERTBIO–UFCG) for optical microscopy and scanning electron microscopy images. NGJ and IDSS thank CNPq for their fellowship. AR thanks CAPES for his post-doctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guimarães Jaques, N., dos Santos Silva, I.D., Ries, A. et al. Nonisothermal crystallization studies of PBT/ZnO compounds. J Therm Anal Calorim 131, 2569–2577 (2018). https://doi.org/10.1007/s10973-017-6754-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6754-x