
Abstract
The calorimetric measurements together with the experiments aimed in the characterization of pore solution and the other analyses relating to the interaction of cement paste with sulphate and nitrate solutions of various concentrations are reported. These salts modify the rate of cement hydration at early age. In the presence of sulphates, the formation of some well-defined calcium sulphates or calcium sulphoaluminates can be found. In case of nitrates, there are no additional products—the nitrate compounds are well soluble, and they do not crystallize in cement systems. However, one can observe that the concentration of ions in the liquid phase is modified and the properties of hydration product are changed, as the formation of products from the liquid phase is disturbed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cement hydration is a heterogeneous process in which the nearly amorphous calcium silicate hydrates are formed, accompanied by crystalline calcium aluminosulphate phase (ettringite—3CaO·Al2O3·3CaSO4·32H2O) and calcium hydroxide. Basic properties of cement pastes are attributed to these calcium silicates, known as C–S–H gel. The formation of hydrated phases leads to the reduction in distance between the grains and the viscosity of paste is enhanced. At the next stage of this process, the rigid skeleton structure is built and this structure reveals ability to transfer some load [1].
When the admixtures in the form of soluble inorganic salts are added to the process water, they affect cement hydration, entering the complex reactions with cement constituents, calcium silicates and calcium aluminate (aluminoferrite). The reactions—nucleation and precipitation of less or more ordered hydration products—occur first of all in the liquid phase (pore solution) which is highly alkaline (almost entirely this is the saturated calcium and alkali hydroxide solution with some concentration of anionic species). The admixtures of salts modify the rate of cement hydration at early age and the composition of hydration products. In the presence of sulphates, the formation of well-crystallized phases—calcium sulphate (gypsum) or calcium sulphoaluminates (ettringite-type, so-called AFt and calcium monosulphate 3CaO·Al2O3·CASO4·(10–12)H2O—AFm phases)—is expected, as it happens during the corrosion process [1]. Ammonium sulphate, as in the case of ammonium salts, acts as acid medium (because of its hydrolysis and partial evacuation of ammonia) and leads to the decalcification of hydrating paste [2]. Consequently, further formation of gypsum is expected. In case of nitrates forming easily soluble components, there are no additional products detected. In some recent works, the formation of solid solutions between the calcium sulphoaluminates and layered double hydroxides corresponding to Ca4Al2(NO3)2(OH)12·4H2O, resulted from thermodynamic modelling, was suggested [3, 4]. However, the role of C–S–H in sorbing nitrates and the other anions has not been elucidated [3, 4]. In concrete technology, calcium nitrate is a well-established concrete admixture mostly used as setting accelerator. Additionally, the compressive strength enhancement has been documented [5, 6]. The studies indicate the reinforcement corrosion and carbonation process mitigation due to the impact on hydrated material, mainly the so-called (C–S–H) calcium silicate hydrate (nearly amorphous) microstructure (modified distribution of porosity). The microstructure of amorphous products and the “history” of bound water consumption are changed in the presence of admixtures used in the experiments.
The hydroxides and salts of alkali metals accelerate the hydration process because of the strong interaction with components of cements. In this case, relatively high equilibrium concentrations of cations stimulate the dissolution of cement phases and finally the decomposition of the main cement constituents (calcium silicates) silicate anionic substructure takes place. The salts of alkali as well as alkali earth metals (calcium, potassium and sodium nitrates; sodium, potassium thiocyanates and sodium carbonates) are the components of commercially available accelerating admixtures to concrete mixture. According to Taylor [1], the accelerating effects of different anions and cations, added in molar equivalent concentrations, on tricalcium silicate hydration in decreasing order can be given as follows:
-
Anions: Br−, Cl− > SCN− > J− > NO3 − > ClO4 −
-
Cations: Ca2+ > Sr2+ > Ba2+ > Li+ > K+ > Na+, Cs+ > Rb+
The development of investigation techniques, particularly the scanning microscopy, allows to characterize the morphological types of nearly amorphous calcium silicate hydrates which constitute up to 80% of paste volume. Depending on the properties of cement, water-to-cement ratio, time of maturing and additives/admixtures used the following classification have been proposed: C–S–H I with the microstructure composed of elongated forms (fibres, rods, needles), C–S–H II with network structure known as honeycomb, composed of plate-like forms, C–S–H III occurring in the form of isometric grains and the C–S–H IV occurring as assemblages of spherical particles, corresponding to the “inner” product formed inside the cement grains and visible under the electron microscope as compact gel [7]. Calcium sulphoaluminate growing from the surface of cement grains to the space between them, soon after mixing with water (up to 1–2 h), occurs in the form of strong fibres; this phase is responsible for the loss of fluidity and further setting of paste [1].
Why do we investigate particularly the role of metal compounds in cement hydration? They are the components of many industrial wastes and residue from the municipal wastes combustion process. Industrial wastes, such as slag and fly ash, have been used for a long time as supplementary raw materials in the production of cement and concrete. On the other side, the reuse of municipal wastes incineration residue is a serious problem because of the presence of some elements (e.g. Cl) and there are many research projects focused in the municipal wastes incineration residue disposal [8, 9]. Metal compounds are immobilized (stabilized) in hydrating cement paste, due to the physical and chemical properties of cement matrix; therefore, a significant reduction in leaching (up to ppm level) is guaranteed [9–12]. The gel-like C–S–H phase of unstable chemical composition and disordered structure is in equilibrium with alkaline pore solution and, as a consequence, the precipitation of hardy soluble compounds (mainly hydroxides) is thus promoted. This C–S–H shows, owing to the high internal surface, very good absorbability [10–12]. Low permeability and porosity (compact microstructure) make the transport of toxic liquids impossible. The replacement and incorporation of foreign ions are promoted by numerous defects in C–S–H structure. All the factors mentioned above are of the synergic character [8].
Heavy metal compounds are not inert as far as the hydration process is concerned. The studies on the effect of heavy metal compounds on cement hydration started many years ago and showed a considerable retardation of setting in many cases [13–16]. Therefore, the solidification/stabilization of the waste is disturbed. It relates particularly to Pb and Zn which have strong retarding effect [14]. This was observed and reported for the first time in sixties of XXc. [15]. Because of the complex character of wastes from one side and the mixtures used in solidification/stabilization from the other side, the effectiveness of the process must be experimentally verified in each case and there is a need to study the mutual relation in some simplified systems.
When the admixtures in the form of soluble inorganic salts are added to the process water, they affect cement hydration, because the concentrations and equilibria in the pore solution are disturbed. This liquid phase itself is highly alkaline (almost entirely this is the saturated calcium hydroxide solution). The ionic species enter the complex reactions with both calcium silicates and calcium aluminate (aluminoferrite). The salts of alkali and alkali earth metals accelerate the hydration process because of the strong interaction with silicate components of cements (dissolution of silicate substructure); simultaneously the calcium ions concentration is suppressed. Some soluble salts with the other accompanying cations, among them the heavy metal ones, particularly those forming the poorly soluble hydroxides, can retard the hydration significantly. In the presence of sulphates not only the crystallization of gypsum or ettringite-type phases is possible but in case of cations formed insoluble sulphates the crystallization of these sulphates too, resulting in serious hydration hindering [18].
The majority of works dedicated to the interaction between the heavy metal compounds and cement mixtures was focused on the determination of the so-called immobilization potential of stabilizing material (through the leaching tests) [e.g. 10, 17]. The microstructure, as one of the factors affecting the process, was not reported frequently [e.g. 17]. However, because in the presence of various salts, the composition of the liquid phase filling the space among the hydrating cement grains is altered, the nucleation and precipitation of the calcium silicate hydrates is generally disturbed. These forms are relatively poorly shaped as compared to those formed in cement paste with no admixtures [11, 17]. The nearly amorphous structure of C–S–H consists, on molecular level, of two substructures: the calcium hydroxide and silicate. In the latter one, different assemblages of silicon–oxygen tetrahedra are randomly associated with the better ordered hydroxide-type substructure. The foreign ions can be included in this structure too [11, 16].
In this study, the impact of sulphates and nitrates on the heat evolution during cement hydration was evaluated and compared. An attempt to explain the results based on the conductometric measurements and microstructural observation takes into account the “state of the art” relating to the hydration process.
Experimental
Materials and methods
The standard Portland cement type CEM I 42 N commercially available was used as a basic component of expansive binders. The specific surface was approximately 3200 cm2 g−1 (as measured by standard Blaine method). The chemical composition is given in Table 1. The following compounds of analytical purity were used to produce the process solutions: (NH4)2SO4, Al2(SO4)3, Na2SO4, MgSO4, Ni(NO3)2, Cd(NO3)2 and Pb(NO3)2.
The microcalorimetry was applied as a basic method for the estimation of hydration progress. The heat of hardening was measured in a differential non-isothermal–non-adiabatic microcalorimeter (of our own laboratory construction, from commercially available elements, equipped with computer-controlled registration and data refinement) on the pastes produced from 5 g cement samples at w/c = 0.5. The starting temperature was 25 °C. The partially restrained expansion of hydrating samples was tested using a special equipment of our own structure. There were the horizontal moulds playing the role of restraining cage with one moving endplate touching the measuring screw joined with the computer-aided registration system (see Fig. 1a). The measurements of electrical conductivity of hydrating cement suspensions were taken using laboratory computer-aided equipment consisting of commercially available waterproof conductivity metre, conductivity sensor, thermometer and magnetic stirrer (see Fig. 1b). Cement suspensions (at w/c from 10 to 100) were doped with admixtures (calculated by mass of cement). The phase composition after 3-day storage in calorimeter was studied by XRD (Philips 1050) for the pastes with admixtures added as 5% by mass of cement (w/c = 0.5) to see the difference between the products formed at early age. The hydration process was stopped by grinding with acetone and repeated washing with acetone followed by drying with cold air. Finally, the fractured, hardened samples, after 3-day storage in calorimeter were studied under the scanning electron microscope (FEI NanoNova 200 with LINK-ISIS EDS microanalyser).

Experimental installations for plastic shrinkage (a) and conductivity (b) measurements
Results
The results are shown as Figs. 2–20 and in Table 2.

Heat of hardening curves for cement pastes with Al2(SO4)3 admixture (w/c = 0.5)

Heat of hardening curves for cement pastes with MgSO4 admixture (w/c = 0.5)

Heat of hardening curves for cement pastes with Al2(SO4)3 admixture (w/c = 0.5)

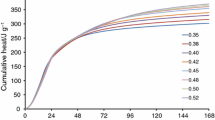
Heat of hardening curves for cement pastes with Na2SO4 admixture (w/c = 0.5)

Heat of hardening curves for cement pastes with 2% nitrate admixtures (w/c = 0.5)

Heat of hardening curves for cement pastes with 5% nitrate admixtures (w/c = 0.5)

Conductivity of cement suspensions processed with Pb(NO3)2, Cd(NO3)2 and Ni(NO3)2 admixtures added as 2% by mass of cement (w/c = 100)

Conductivity of cement suspensions processed with MgSO4, (NH4)2SO4 and Al2(SO4)3 admixtures added as 2% by mass of cement (w/c = 10)

Conductivity of cement suspensions processed with water and with 2% Na2SO4 (w/c = 10); see the scale in Fig. 8

Shrinkage of pastes produced from laboratory made cement hydrated with 2% Cd(NO3)2, Pb(NO3)2 and Ni(NO3)2 solutions (w/c = 0.5)

XRD pattern of hydrated cement samples admixtured with 5% sulphate solutions (w/c = 0.5), after 3-day storage in calorimeter; abbreviations: Gy gypsum, Ett ettringite, CH calcium hydroxide. 1 water; 2 Na2SO4; 3 MgSO4; 4 (NH4)2SO4; 5 Al2(SO4)3

Microstructure of cement paste with no admixture, after 3-day storage in calorimeter—typical microstructure of ettringite (long needles) and calcium silicate hydrates (short fibres, small particles forming “honey-comb” structure)

Microstructure of cement paste processed with 2% NiNO3 solution, after 3-day storage in calorimeter; see very poorly shaped, compact C–S–H

Microstructure of cement paste processed with 2% Cd(NO3)2 solution, after 3-day storage in calorimeter; see very poorly shaped, compact C–S–H

Microstructure of cement paste processed with 2% Pb(NO3)2 solution, after 3-day storage in calorimeter; see poorly shaped compact C–S–H and calcium hydroxide plate-like crystals

Microstructure of cement paste processed with 2% Al2(SO4)3 solution, after 3-day storage in calorimeter; see well-developed ettringite elongated crystals

Microstructure of cement paste processed with 2% (NH4)2SO4 solution, after 3-day storage in calorimeter; see well-developed gypsum crystals

Microstructure of cement paste processed with 2% Na2SO4 solution, after 3-day storage in calorimeter; see well-developed forms of various hydrated products

Microstructure of cement paste processed with 2% MgSO4 solution, after 3-day storage in calorimeter; see well-developed plate-like gypsum crystals
As it is commonly known in cement chemistry, only the calorimetric measurements give the possibility to follow the changes occurring in the hydrating mixture produced at standard water-to-cement ratio in a continuous way from the beginning of the process, without removal of water (e.g. for the XRD or DTA studies the so-called chemical drying should be done and the data obtained vs. time are of discrete character). The heat-evolved versus time curves reflect very precisely the progress of complex phenomena and reactions with water, accompanying the increasing content of hydration product. After the initial intensive heat evolution during the first minutes (water adsorption and surface dissolution), the heat evolution decreases in so-called dormant period and subsequently the heat evolution (corresponding to the growth of hydration products) increases steadily and a maximum is attained; the increase in total heat-evolved values becomes very slow after 2–3 days when the layer of products is formed on cement grains and the process is diffusion controlled [e.g. 1]. According to the standards, the values after 41 h from the processing with water are measured and evaluated. Every retarding admixture brings about the reduction in heat-evolved value that increases throughout some period of time at early age of hardening (it means even up to 2 days). The retardation of hydration is observed particularly when the hydration takes place in the presence of Pb compounds [see also 15, 18].
The calorimetric curves (Figs. 2–7) reflect well the effects attributed to the action of sulphate, nitrate anions and accompanying cations.
As one can see in Figs. 2–4 the heat evolution/hydration process in the presence of aluminium sulphates and ammonium and magnesium sulphates strongly depends on the concentration of admixture. This is particularly well visible in the case of aluminium sulphate. At 2% admixture, the shape of calorimetric curve seems to indicate the formation of sulphoaluminate product (sharp peak), presumably the ettringite phase. At 5% the prolonged induction period results presumably from the precipitation of ettringite on the hydrating grains; however, after 15 h the intensive hydration is continued. At 10% the heat evolution is untypical—the low, sharp peak corresponds presumably to the formation of thick ettringite layer, hampering further hydration. The heat-evolved data correspond well with these results. At MgSO4 admixture, a prolonged up to 5-h induction period means retardation at early age (Fig. 3); one can presume that soluble admixture brings about an initial slow nucleation of hydration products and the crystallization of some gypsum amount is possible. However, an intensive hydration occurs, as it has been derived from the heat-evolved data (Table 2). At 5% concentration the heat evolution/hydration process is “broadened” over longer period of time and the heat values are similar to those for reference cement paste.
Ammonium sulphate brings about the some retardation (Fig. 4) at early age, more evident at higher concentration; the heat-evolved data correspond well with the plots (Table 2).
In turn, the sodium sulphate gives markedly accelerating effect—the increase in total heat-evolved values with concentration of admixture (from 2 to 5%) is observed and these heat values are the highest (Table 2). There is not heat evolution curve shape modification—only the main heat evolution peak becomes higher and the induction period is not shortened. It seems that the mechanism of hydration products formation is not specially changed; however, because of intensive dissolution of calcium silicate cement component, growing with concentration the decomposition process is more effective (Fig. 5).
A slightly accelerating effect of nickel and cadmium nitrate is well visible at 2% concentration (Fig. 6); the main peaks are slightly moved toward the beginning of hydration and at later age, the heat evolution curves do not decline specially from that for the reference. Some more evident change of heat evolution curve is observed at 5% concentration; however. the heat values (Table 2) are not specially altered as compared to reference. It means that these metals containing sludge which should be stabilized/solidified will undergo “normal” setting process. Simultaneously, it is clearly seen that the hydration process is strongly retarded in the presence of lead compound (PbNO3) irrespectively of concentration (Figs. 6 and 7; Table 2). The so-called dormant period is extended; it means that in these pastes the content of hydration products is low and the renewed hydration occurs after almost 40 h.
The calorimetric data correspond well with the conductivity measurements results. Of course, because of practical reasons, the water-to-cement ratio 0, 5, as in calorimetric studies, cannot be applied. However, because of very low solubility of cement constituent and hydration products (mmol dm−3), the rate of liquid phase saturation and precipitation of products does not decline markedly when this ratio is 0, 5; 10 or even 100, respectively. In spite of this discrepancy, the relationships between the hydrating samples in particular series can be indicated and the action of admixtures can be compared. One should underline that this method reflects the changes of concentration of ions present in the solution versus time. It means the difference between the dissolution and precipitation process. In neat cement suspension, there is a sharp peak corresponding to the supersaturation state, after which the rapid nucleation occurs and the formation of products takes place (see Figs. 8, 9). The same shape of conductivity curve is observed at the presence of nitrates. In case of magnesium, ammonium and aluminium sulphate-admixtured samples, the lowering of conductivity, resulting from the precipitation of hardly soluble sulphate salts, is significant (Fig. 9), and there is no peak visible. The shortage of concentration peak can be attributed to the rapid precipitation of sulphate products. The effect of gypsum crystallization (see XRD pattern in Fig. 12) results in similar conductivity decrease in the ammonium and magnesium sulphate containing suspensions. An intensive ettringite crystallization in the presence of Al2(SO4)3 (see XRD data, Fig. 12) corresponds well to the lowest conductivity of hydrating sample (Fig. 9). On the other side, in cement suspension enriched with sodium sulphate, the liquid phase is rich in ions derived both from soluble admixture and from dissolved cement phases (see both Figs. 9, 10); there is neither gypsum nor ettringite present in significant amount as it can be deduced from XRD data.
The amount of ettringite is the highest in the presence of Al2(SO4)3 admixture (Fig. 12) though this product is detectable in the other hydrating samples. The formation of this calcium sulphoaluminate phase is particularly well illustrated by SEM images. In the sample admixtured with aluminium sulphate (Fig. 17) and in the other samples (Figs. 13, 18, 19), well-developed ettringite crystals (laths, rods) are accompanied by less developed or poorly shaped C–S–H. One can find gypsum crystals in the pastes processed with MgSO4 and (NH4)2SO4 solutions (Figs. 18, 20).
In the presence of nitrates, the formation of poorly shaped calcium silicate hydrate phase was generally found (Figs. 14–16). This product shows various water content; as it results from the changes of paste volume (see Fig. 11). C–S–H reveals, as compared to the reference sample, significant plastic shrinkage in the presence of Ni(NO3)2 or even slight swelling in the presence of Cd(NO3)2. One can speculate that the hardened binder structure formed in the presence of these compounds should be compact, more impermeable and more susceptible to the attack of external aggressive solutions. The retarding effect of Pb (NO3)2, very well seen on the calorimetric curves (Figs. 6, 7), can be proved too by shrinkage measurements—the changes of volume, as compared to the other samples, are negligible.
Conclusions
-
The rate of heat evolution during hydration (equivalent to the hydration kinetics), as well as the composition of hydration products and microstructure, is strongly affected by sulphates and nitrates.
-
The total heat evolution, equivalent to the progress of hydration products formation, reflects indirectly the kinetics of dissolution/precipitation reactions, occurring in the liquid phase, enriched with soluble admixtures. At low concentration (2%) Mg, ammonium, Al sulphate, as well Cd, Ni nitrates act as hydration accelerators.
-
The hydration process is strongly retarded in the presence of Pb nitrate and strongly accelerated in the presence of sodium sulphate.
-
The hydration process is retarded in 5% solution of ammonium sulphates; the retarding effect of more concentrated nitrates is not significant.
-
The high amount of needle-like ettringite is observed in the presence of aluminium sulphate; gypsum is observed too as a result of calcium ions reaction with sulphate; this takes place particularly in case of ammonium and magnesium sulphate. The formation of gypsum or ettringite was proved by XRD.
-
At the presence of metal compounds, the formation of calcium silicate well-shaped forms is disturbed. This is particularly evident in the presence of nickel and cadmium nitrate. Therefore, the structure becomes more compact and impermeable.
References
Taylor HFW. Cement chemistry. London: Thomas Telford Publishing; 1997.
Varga C, Alonso MM, Mejía de Gutierrez R, Mejía J, Puertas F. Decalcification of alkali-activated slag pastes. Effect of the chemical composition of the slag. Mater Struct. 2015;48:541–55.
Balonis M. The influence of inorganic chemical accelerators and corrosion inhibitors on the mineralogy of hydrated cement systems, PhD thesis, University of Aberdeen; 2010.
Balonis M, Mędala M, Glasser FP. Influence of calcium nitrate and nitrite on the constitution of AFm and AFt cement hydrates. Advances in Cement Research. 2011;23(3):129–43.
Justnes H, Franke W. Calcium nitrate as multi-functional concreto admixtures, no 3, 50–53, Budownictwo, Technologie, Architektura; 2015.
Franke W, Weger D, Skarabis J, Gehlen Ch. Study on calcium nitrate impact on carbonation of concrete. In: 1st international conference on grand challenges in construction materials, Los Angeles; 2016.
Diamond S. In: Hydraulic cement pastes: their structure and properties. In: Proceedings of the conference, cement and concrete asso, Wexham Springs; 1976.
Glasser FP. Immobilisation potential of cementitious materials, environmental aspects of construction with waste material. Amsterdam: Elsevier; 1994. p. 77–86.
Nocuń-Wczelik W, Trybalska B, Rakowska A. The microstructure of C–S–H formed in the presence of heavy metal compounds, Polski Biuletyn Ceramiczny Nr 8, Ceramika 46, Kraków; 1994.
Glasser FP. Application of cements to the treatment and conditioning of toxic wastes. In: 9th International congress on the chemistry of cement, New Delhi. India; 1992. p. 114–18.
Nocuń-Wczelik W. Struktura i właściwości uwodnionych krzemianów wapniowych, Prace Komisji Nauk Ceramicznych, Polski Biuletyn Ceramiczny Nr 18, Ceramika 59, Kraków; 1999.
Nocuń-Wczelik W. Immobilizacja metali ciężkich przez fazę C–S–H, Cement-Wapno-Beton, II/LXIV; 1997. p. 188–91.
Rossetti VA, Medici F. Inertization of toxic metals in cement matrices: effects on hydration, setting and hardening. Cem Concr Res. 1995;25:1147–52.
Fernandez-Olmo I, Chacon E, Irabien A. Influence of lead, zinc, iron(III) and chromium(III) oxides on the setting time and strength development of Portland cement. Cem Concr Res. 2001;31:1213–9.
Lieber W. The influence of lead and zinc compounds on the hydration of Portland cement. In: 5th International symposium on the chemistry of cement, Tokyo, vol. 2; 1968. p. 444–54.
Gineys N, Aouad G, Damidot D. Managing trace elements in Portland cement—part I: interactions between cement paste and heavy metals added during mixing as soluble salts. Cem Concr Compos. 2010;32:563–70.
Mellado A, Borrachero MV, Soriano L, Paya J, Monzo J. Immobilization of Zn(II) in Portland cement pastes. Determination of microstructure and leaching performance, J Therm Anal Calorim. 2013;112:1377–89.
Nocuń-Wczelik W, Łój G. Effect of lead compounds on alite hydration. Cement-Wapno-Beton, XI/LXXIII. 2006;6(343–350):285–90.
Acknowledgements
The financial support from the Faculty of Material Science and Ceramics, University of Science and Technology AGH in Cracow, Poland is greatly acknowledged (Grant No 11.11.160.415).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nocuń-Wczelik, W. Differential calorimetry as a tool in the studies of cement hydration kinetics with sulphate and nitrate solutions. J Therm Anal Calorim 130, 249–259 (2017). https://doi.org/10.1007/s10973-017-6378-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6378-1