
Abstract
The miscibility, crystallization and melting behaviour of poly(trimethylene terephthalate)/thermotropic liquid crystalline polymer (LCP) blends were studied using differential scanning calorimetry. The blends were found to form primarily an immiscible system. The addition of LCP accelerated the overall rate of crystallization and caused a depression in equilibrium melting temperature, especially at low LCP content. Lauritzen–Hoffman analysis showed that the addition of LCP caused a reduction in the fold surface energy and increased the regime II to III transition temperature.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
LCP-based blends have been the focus of attention of several studies [1–5]. Due to their ‘rigid rod’ type structure and consequent long relaxation times, when LCP molecules are sheared above its solid to nematic transition, they align themselves in the direction of flow. Thus, when they are added in a minor phase, they generally tend to improve the processability of the polymer matrix. In addition, depending on the viscosity ratio, the LCP droplets may deform and elongate under extensional flow, thus undergoing in situ fibrillation and enhancing the mechanical properties of the polymer matrix [1–6].
The crystallization behaviour is another important factor in the processing of polymer blends. The crystallization process due to its resultant volume contraction can induce stresses which freeze in during solidification and can thus affect the quality of the final product [6]. Factors such as crystalline or amorphous nature of the minor phase, component concentration, relative melt viscosities and miscibility can greatly affect the crystallization behaviour of polymer blends. In this regard, several studies have been carried out on the crystallization kinetics of LCP-based polymer blends [7–18]. There are reports that in the case of immiscible blends, the LCP minor phase accelerates the crystallization process in its blends with poly(butylene terephthalate) (PBT) [7, 9], poly(phenylene sulphide) (PPS) [8, 10, 17, 19, 20], polypropylene, maleic anhydride grafted polypropylene [11, 21], poly(ethylene naphthalate) (PEN) [12, 18] and poly(ethylene terephthalate) (PET) [13]. However, in other cases, it was found to decrease the crystallization rate in PET [22] and PPS [23]. In the case of blends of poly(ether ether ketone) (PEEK) with Vectra LCP, it was found to slightly increase the crystallization rates in the isothermal studies [16], while decrease the rates in the nonisothermal case [15]. de Carvalho and Betras [14] for PEEK/HX4000 LCP blends found that the addition of LCP decreases the overall crystallization rate. The authors suspected partial miscibility between the components.
In miscible crystalline/amorphous systems, surface nucleation rate depends on the rate of attachment of crystallizable segments onto the growth front as well as the rate of diffusion of amorphous segments away from the growing crystal [24]. In partially miscible systems, complex behaviour can develop due to the competitive effects of nucleation and dilution by the minor phase. Lee et al. [25] found that polycarbonate (PC) forms a miscible system with LCP at very low concentrations and a partially miscible system for the rest. Semiflexible LCP based on PET and p-amino benzoic acid forms a miscible blend with Nylon 66 [26] in the melt state and a partially miscible system in the solid state. The authors reported that blending causes a depression in the equilibrium melting temperature as well as in the rate of crystallization of the matrix. Xie et al. [27] found Nylon 66/LCP blends to be miscible. They also reported PBT/LCP blends to be miscible up to 10 mass% of LCP. Zhong et al. [28] found PEEK/LCP blends to be partially miscible based on a single composition-dependent T g. The LCP used therein was a copolyester of p-hydroxybenzoic acid, terephthalic acid and resorcinol. The addition of LCP caused a decrease in the melting temperature; however, it increased the crystallization rate and the degree of crystallinity in the blends.
In the light of contrary results reported listed above, it appears that the crystallization behaviour in LCP-based blends is not fully understood. Furthermore, miscibility appears to play vital role in governing the crystallization behaviour in addition to other factors such as processing parameters and thermal history. The current study will examine the miscibility and crystallization behaviour of PTT/Vectra A950 LCP blends. There have been reports [29, 30] on the nonisothermal crystallization studies of PTT/LCP blends. The authors in these cases have reported that the PTT/LCP blends form an immiscible system where the minor component promoted greater rates of crystallization. The current study will seek to re-examine the issue of miscibility of PTT/LCP blends and study its effects on the bulk crystallization processes under isothermal conditions.
Theoretical background
Crystallization kinetics studies can involve quantification of bulk crystallization rate using a macrokinetic model such as Avrami [31–33] and/or study of the growth of the initially formed nucleus using surface nucleation theory proposed by Hoffman et al. [34].
Bulk crystallization studies
In isothermal crystallization studies, the relative crystallinity at a particular time t can be obtained from the heat flow curves using the following relation [35]:
where t = 0 corresponds to beginning of the crystallization curve and t = ∞ corresponds to the end of the crystallization curve. Using the χ(t) data, the bulk crystallization kinetics can described by a macrokinetic model proposed by Avrami of the form [31–33]:
where χ(t) is the relative crystallinity, k is the Avrami rate constant, and n is the Avrami exponent. Taking double logarithms, it can be expressed as:
By plotting \(\log \left[ { - \ln \left\{ {1 - \chi \left( t \right)} \right\}} \right]\) against log t, the Avrami parameters, i.e. n and k, can be obtained from the slope and intercept, respectively.
Surface nucleation theory
The Lauritzen–Hoffman (L–H) theory predicts, during crystallization, three distinct growth regimes, i.e. regimes I, II and III, that occur at progressively lower temperatures. According to this theory, the growth of initially formed primary nucleus through surface nucleation can be expressed by the relation [34]:
The term G represents the spherulite growth rate; G 0 is the temperature-independent pre-exponential factor. The second exponential term accounts for the energy required for the formation of critical nucleus. U* represents the activation energy for transport of polymer segments to the liquid–crystal interface. R is the universal gas constant. T ∞ is the temperature at which viscous flow ceases and is usually taken to be (T g–30 K). The term f is a correction factor to account for temperature dependence of enthalpy of fusion and is denoted by f = (2T c/T c + T 0m ). It has values close to unity.
In the above relation, ΔT represents the undercooling, i.e. T 0m − T c, where T 0m and T c represent the equilibrium melting temperature and crystallization temperature, respectively. The equilibrium melting temperature represents the melting temperature of an infinitely thick crystal where the chemical potential of the amorphous liquid and crystalline components is equal [36]. The Hoffman–Weeks plot is used to calculate T 0m through the following relation [37, 38]:
where T m represents the melting temperature of crystalline lamella crystallized at T c, γ represents the thickening factor, i.e. the ratio of final lamellar thickness (l) to the initial lamellar thickness (l*). The intersection point of T m = T c line with the T m versus T c line would yield \(T_{\text{m}}^{0}\).
In Eq. (4), K g represents the nucleation parameter described by the following relation:
In the above relation, b represents the monomolecular layer thickness. σ and σ e represent the lateral and the fold surface energies, respectively. ΔH f is the enthalpy of fusion per unit volume. k b is the Boltzmann constant. z is the regime parameter and has a value of 4 for regimes I and III and 2 for regime II.
Regime I growth occurs at the highest temperatures wherein a single surface nucleus causes completion of substrate of length L. The overall growth rate corresponds to G = biL [34], where i is the surface nucleation rate. The spherulitic growth occurs through the addition of a layer of substrate of thickness b. At lower temperatures, in regime II, due to an increase in the surface nucleation rate, multiple nuclei attach themselves to the substrate at a rate comparable with the substrate completion rate (g). The overall growth rate in this second regime can be expressed with G = b(2ig)1/2.. At larger undercooling (regime III), due to much larger increase in the surface nucleation rate, the overall growth rate once again corresponds to G = biL. Consequently, in the L–H plots obtained from Eq. (4), regimes I and III have a slope twice of that in regime II.
Experimental
Materials
PTT (Corterra 9200) pellets were donated by Shell chemicals (USA). Vectra A950 LCP which is a random copolymer of p-hydroxybenzoic acid and 2,6- hydroxynaphthoic acid in the ratio 73/27 was gifted by Ticona (USA). The blends were prepared by melt mixing in Haake minilab I. The Haake miniblender consists of a conical twin screw arrangement with a back-flow channel. The samples were melt-mixed at 290 °C at 80 rpm after initial loading at 50 rpm. The total residence time was set at 3 min. Pure PTT, LCP and its blends in 90/10, 80/20, 70/30, 60/40 compositions were prepared using the previously mentioned parameters. The samples after extrusion were dried in an oven at 110 °C for 4 h and subsequently used for DSC characterization.
Methods
Differential scanning calorimetry (DSC)
The miscibility and crystallization kinetics studies were carried out on Perkin Elmer DSC 8000 and DSC 7 calorimeters, respectively. The temperature and heat flow of both DSCs were calibrated with an indium standard. The onset (156.6 °C) and enthalpy of fusion (28.45 J g−1) were used for calibration. The calibration was periodically checked to ensure reliability of the data. The sample mass was kept constant at 4 mg.
The temperature program for miscibility experiments involved heating and cooling at a scan rate of 60 °C min−1 from 10 to 300 °C, holding at the end temperature for 5 min in order to eliminate the thermal history. Subsequently, the T g was determined from the second run at a scan rate 40 °C min−1.
The isothermal crystallization studies involved heating the sample from 50 to 300 °C at 60 °C min−1, holding at the end temperature for 5 min to erase the thermal history and rapidly cooling at 160 °C min−1 to the desired isothermal temperature. The sample was kept at the crystallization temperature for 60 min to ensure completion of crystallization. The isothermal crystallization temperatures selected were 178, 180, 182, 184, 186, 188, 190, 192, 194, 195, 196, 197, 198, 199, 200 and 201 °C. The experiments were carried out under a nitrogen atmosphere at a constant flow to prevent thermal degradation.
Scanning electron microscopy
The samples extruded from the miniblender were cryofractured and sputter-coated with gold prior to imaging. The phase morphology was studied using a JEOL JSM 6380LA analytical microscope. Initially, for the 70/30 composition, 20 kV accelerating voltage was used. However, due to sample charging, the voltage was reduced to 10 kV for the other samples.
Polarized light microscopy
The crystalline spherulitic morphology of isothermally crystallized samples was characterized by an Olympus BX50 microscope with a 4× objective. The samples were prepared by melting them on a hot plate in between a glass slide and a cover slip at 300 °C for 5 min and then transferring them to the heating stage of a Haake rheometer. The temperature was set to 200 °C, where it was crystallized for 1 h. The spherulite images were captured with a Logitech webcam which was fitted to the microscope. Carl Zeiss Axiovision software was used to generate appropriate scale bars for the objective from the image of a stage micrometer.
Results and discussion
Miscibility studies
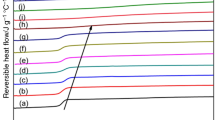
Figure 1a displays the heat flow curves from the second heating runs. The blends display a single composition-dependent T g. The high heating rate was chosen to increase the heat signal, yet the T g corresponding to the LCP component in the blends could not be determined. However, a small peak corresponding to the melting of LCP crystals can be seen in the high-temperature region. In the case of pure LCP heating curve, the low-temperature melting peak may arise out of crystal perfection [39]. The results displayed in Table 1 show an increase in the value of the T g of the blends with LCP content up to the 80/20 composition, decreasing thereafter, thus indicating the possibility of miscibility in the amorphous phase at low LCP concentrations.

Heat flow curves depicting a second heating, b first cooling curves of PTT, LCP and its blends. The melting peak of LCP can be seen in the 260–280 °C temperature range
The results displayed also indicate that the melting temperature (T m) of PTT and LCP component is marginally lower in the blend. This behaviour will be examined in detail in the melting experiments after isothermal crystallization. The presence of separate melting peaks rules out the possibility of formation of cocrystals, as expected, due to differences in their chemical structures [40]. Figure 1b depicts the first cooling curves. Whereas the crystallization peak temperature (T p) of the PTT component in the blends is higher than that of pure PTT, the T p of the LCP component is lower than that of pure LCP, especially for the 90/10, 80/20 compositions.
In order to offset the effect of initial crystallinity on the results, additional runs were carried out on the samples. Fresh samples of similar mass, encapsulated in aluminium pans, were allowed to melt on a hot plate at 300 °C for 5 min and quenched in liquid nitrogen (LN2). The samples were subsequently transferred to the DSC 8000 sample holder, and heating runs from 20 to 300 °C were carried out at 10 °C min−1. The corresponding heat flow curves displayed in Fig. 2 show well-defined T gs and cold crystallization peaks reflecting the amorphous nature of the samples. The T g of the PTT component, in this case, displays only a marginal shift to higher temperatures. The shift was also observed in additional experiments carried out at different scan rates. The cold crystallization peak temperature also displays a marginal shift above the pure PTT value for the 70/30, 60/40 compositions.

Heat flow curves of PTT and its blends after LN2 quenching
The SEM images of the blends displayed in Fig. 3a–d show a two-phase system, with the samples, especially at higher LCP concentrations, and exhibit a typical droplet–matrix system. At high concentrations of LCP, a larger droplet size results from phase separation and coalescence of the minor phase, as typically seen in immiscible systems [8]. The presence of a diffused interface observed for the 90/10, 80/20 compositions may be indicative of some interaction between the components. Pishrath et al. [29] reported SEM images where the components exhibited sharp interfaces with voids for similar compositions. The samples in those cases were processed at lower temperatures where there is lesser possibility of interaction between the components though the process of transesterification. However, in the current system, as noted from the marginal shift in the T g results, the high processing temperature does not appear to induce any significant level of miscibility and the system appears to be primarily an immiscible two-phase system.

Scanning electron micrographs of the PTT/LCP blends in a 90/10, b 80/20, c 70/30 and d 60/40 compositions
Furthermore, it appears that for the melt crystallization case, on cooling from the melt, LCP-rich domains crystallize first in the presence of molten PTT matrix. The presence of such a flexible molten domain causes an apparent reduction in T p(LCP). The crystalline LCP phase subsequently nucleates the PTT which increases the T p(PTT) values (Table 1). For the LN2-quenched blend samples, especially for the 90/10, 80/20 compositions, this nucleating effect in the blends is diminished due to suppression of the crystallization of the LCP phase. We have carried out additional cold crystallization studies where the initial level of crystallinity in the samples was increased by allowing it to cool in the DSC cell itself. The minor phase in those cases exhibited greater nucleating effect and caused a larger shift of cold crystallization peak of the blends towards lower temperatures. It should be noted that in the cold crystallization case, although the nucleating effect of the crystalline domains is reduced, there might be some degree of domain interface as well as impurity driven nucleation of the matrix [41].
Crystallization behaviour
The crystallization curves of PTT and its blends for a particular isothermal temperature (T c) are displayed in Fig. 4a. For the blends, the curves tended to become narrower and showed decrease in area with the peak shifting to lower times with an increase in LCP content.

a Normalized heat flow curves, b plots of relative crystallinity versus time, c Avrami plots of PTT and its blends at T c 196 °C
A representative plot of relative crystallinity versus time using Eq. (1) is shown in Fig. 4b. The area calculations were carried out using the Perkin Elmer Pyris software, with a straight baseline, according to procedure suggested by Lorenzo et al. [42].
The curves exhibit a typical sigmoidal-shaped nature, moving to a shorter timescale with increasing content of LCP indicating early completion of the crystallization process. The nonlinear nature of the curves at the beginning and at the end relates to the induction period and the secondary crystallization process, respectively. The induction period corresponds to the time required to achieve steady-state nucleation [43].
Figure 4c displays a typical Avrami plot for PTT and its blends obtained using Eq. (2). The Avrami equation is used to describe the primary crystallization process until impingement. Hence, only the linear portion of the plots corresponding to 10–80% conversion was used for application of the model [44]. It can also be seen from Fig. 4b that the conversion range corresponds to linear portion of the χ(t) plots The obtained plots were highly linear, as seen from the value of correlation factor (r 2) listed in Table 2. The values of k and n obtained from such plots along with other parameters are shown in Table 2. The other parameters listed include the half-time of crystallization, i.e. time taken for 50% conversion (t 0.5), the induction time (t 0.1), i.e. time taken for 10% conversion, that were obtained from the plots of χ(t) and the width of crystallization exotherm (Δw).
The values of t 0.5 and k are used to quantify the bulk crystallization rate. For a particular composition, t 0.1 and t 0.5 decrease with reduction in T c. The lowering of the free energy barrier to nucleation with the reduction in T c can promote high primary nucleation rates and consequently raise the overall bulk crystallization rates. This effect can also be seen through the higher values of k at low T cs. Δw also decreases with reduction in T c. Moreover, in the blends, Δw mostly decreases with increasing LCP content, signifying early completion of the crystallization process. The values of t 0.1 and t 0.5 show a similar decrease for the blends up to the 70/30 composition increasing slightly for the 60/40 composition. The current results thus confirm the previous conclusion that the LCP crystalline phase domains nucleate the PTT matrix and promote higher crystallization rates. The LCP crystalline domains by acting as heterogeneous impurities and providing surfaces reduce the free energy barrier to nucleation and therefore promote high primary nucleation rates. This leads to lower induction times and consequently lower t 0.5 as well as an increase in k in the blends as compared to PTT. In the blends with high LCP content, especially for the 60/40 composition, the agglomeration of LCP domains results in diminished nucleating efficiency by reducing the number of sites available for growth. This can lead to lower bulk crystallization rates for those compositions, although it remains higher than PTT.
The Avrami exponent (n) depends on the nature of generation of primary nuclei, i.e. homogeneous or heterogeneous, as well as the dimension of the growing crystal. The ‘n’ values for the blends remained mostly below that of PTT due to heterogeneous nucleation as seen from the optical micrographs. For a particular composition, ‘n’ tends to increase with the rise in T c up to 190 °C decreasing slightly thereafter. ‘n’ values for most of the compositions remain between 2 and 3 which correspond to three-dimensional spherulitic growth preceded by heterogeneous nucleation. The crystalline morphology of the samples might change from truncated spherulitic to more spherulitic with the increase in T c. In addition, the higher temperatures may promote sporadic or thermal generation of nuclei [45] which may explain the higher ‘n’ values.
The enthalpy of crystallization (∆H c) increases with the rise in T c indicating higher levels of crystallinity. At the higher T cs, thicker lamellar crystals are developed to attain stability and thus promote an overall increase in the level of crystallinity. In the blends, ∆H c decreases with increasing LCP content due to lowering of PTT concentration in the composition. LCP, due to its low melt entropy, exhibits a very low enthalpy of crystallization, which cannot be easily detected by DSC [6]. Thus, the contribution to the enthalpy can be entirely attributed to PTT. Furthermore, the normalized enthalpy of crystallization (∆H c(normalized)) decreases in all compositions as compared to PTT, despite an increase in the crystallization rate. However, in the blends with low LCP content, ∆H c(normalized) is very close to PTT. A similar observation was made in LCP-based blends and was thought to be the result of minor phase promoting the formation of imperfect crystals of the matrix component [13, 30, 46].
Crystalline morphology
Figure 5a–e displays the optical micrographs of PTT and its blends crystallized at 200 °C. As the concentration of LCP is increased, the nucleation effect of the minor phase can be seen through the increase in the spherulite density and formation of smaller spherulites. For the 60/40 composition, the spherulite size increases slightly. The spherulites show a clear Maltese cross caused by alignment of chain axis with the crossed polarizers. A banded structure [47, 48] reported for PTT for these temperatures is not present. The micrographs of pure PTT as well as for 90/10 composition display sharp boundaries which is a characteristic of heterogeneously generated nuclei [49]. The grainy structure can be attributed to the LCP nematic component. In the blends with low LCP content, the PTT spherulites are space filling indicating that the minor component is incorporated within the spherulites in interfibrillar regions [50]. Thus, in the blends with low LCP concentrations (Fig. 5b, c), due to reduced mobility, as seen from the T g results, and more significantly, due to increase in growth rate that can be estimated from t0.5 values (see later sections), the noncrystallizable LCP phase remains trapped in the interfibrillar region [36, 50–53]. At high LCP concentrations (Fig. 5d, e), due to reduced growth rate, it appears to be rejected primarily in the interspherulitic region.

Polarized light microscope photograph of PTT/LCP blends in a 100/0, b 90/10, c 80/20, d 70/30, e 60/40 compositions after isothermal crystallization at 200 °C for 1 h
Melting behaviour
Figure 6a displays the subsequent melting behaviour of the PTT crystallized at different T cs. According to Srimoaon et al. [54], peaks I and II are formed due to melting of secondary and primary crystallites, respectively. Peak III might have formed due to partial melting of the less stable fraction of primary as well as secondary crystals, and peak IV is attributed to melting of crystals formed out reorganization of more stable fraction of the primary crystals during heating. The first and second melting endotherm (peak II) increases in intensity and shifts to higher temperatures with an increase in T c. In addition, the third melting peak (peak III) tends to merge with the fourth peak forming a single peak above T c–194 °C. A small recrystallization exotherm can be seen at the beginning of the third melting peak for samples crystallized at low T cs which decreases in intensity at high T cs. The highest temperature peak (peak IV) generally shows no significant variation in position with T c. It should be noted here that the peak corresponding to the LCP component could not be determined. It may be the result of low level of sensitivity of DSC 7 instrument; also, it may reflect the low degree of crystallinity of LCP phase.

Melting behaviour of a PTT after isothermal crystallization at different T cs. b PTT and its blends at crystallized T c 200 °C
In the case of blends, as depicted in Fig. 6b, the inclusion of LCP, especially at low concentrations and at high T cs, has appeared to marginally lower the peak temperature corresponding to primary crystals, thus indicating the formation of thinner imperfect crystals. The peak temperature corresponding to melting of primary crystals can be further used to determine the equilibrium melting temperature. Table 3 displays the values of \(T_{\text{m}}^{0}\) and γ for PTT and its blends calculated using Eq. (5). The T m values corresponding to the melting of primary crystallites, i.e. peak II, were used to obtain the Hoffman–Weeks plots. The Hoffman–Weeks plot assumes a constant γ in the crystallization range. Nonconstant values of γ can lead to deviations from the straight-line behaviour at high T cs. At low T cs, additional terms must be added to Eq. 5 which leads to a near constant dependence of T m on T c [37]. Hence, the values of \(T_{\text{m}}^{0}\) can vary depending on the T c range selected. In the present case, the T c range of 188–201 °C was selected. The value for the \(T_{\text{m}}^{0}\) obtained for PTT is close to the value (248.2 °C) obtained by Supaphol et al. [55] in a similar T c range. γ does not significantly vary for the blends, indicating that the initially formed thin lamellae underwent similar degree of thickening to achieve stability during isothermal crystallization. Furthermore, for the blends, \(T_{\text{m}}^{0}\) decreases with increase in LCP content up to 80/20 composition, increasing thereafter for 70/30, 60/40 compositions. This depression of \(T_{\text{m}}^{0}\) can be a consequence of thermodynamic or kinetic considerations [56]. According to the kinetic, morphological approach [57], the depression in \(T_{\text{m}}^{0}\) can be a consequence of changes in crystal perfection.
Lauritzen–Hoffman analysis
Equation (4) can be modified to express the spherulite growth rate in terms of the half-time of crystallization (t 0.5) [58, 59]:
where \(\left( {\frac{1}{{t_{0.5} }}} \right)_{0}\) is the temperature in independent pre-exponential factor.
Taking logarithms and rearranging we get the Lauritzen–Hoffman plots:
In order to test the applicability of the above approximation, Achilias et al. [60] conducted studies in which the time corresponding to different conversions as well as U* values was substituted in Eq. (8) and ratio of the slopes of the L–H plots was noted. The studies were carried out on PTT of different molecular weights. The authors found that the time corresponding to 2% conversion gave the optimal results in terms of the ratio of the slopes between regimes III and II being closest to the theoretical value of 2.
In the current study, we found that the values corresponding to 10% conversion gave the optimal results. At lower conversions, a scattering of data points was observed, especially at higher T cs.
The L–H plots in Fig. 7 were obtained using U* = 4120 cal mol−1 and T ∞ = T g − 51.6 K instead of the ‘universal’ values of 1500 cal mol−1 and T ∞ = T g − 30 K [34], respectively. The L–H plot for PTT using half-time of crystallization is also displayed for comparison. The nucleation constants were obtained from the slopes of the curves, and the product σσ e is displayed in Table 4. The ratio of the slopes obtained from using t 0.5 values is lower than that obtained from t 0.1 values, thus supporting the decision to use t 0.1 values. The K g values for the blends, obtained from the slope, decrease up to the 80/20 composition and subsequently increase for the other compositions. The product σσ e obtained from Eq. (6) follows a similar trend. The low K g through reduced σσ e values indicate a diminution in the free energy barrier to nucleation for those compositions. At a particular undercooling, such a decrease in K g would promote an increase in the overall growth rate for those compositions. This can be seen from the plots of 1/t 0.1 in terms of ΔT displayed in Fig. 8. It can be observed that despite the reduced driving force through a decrease in \(T_{\text{m}}^{0}\), the blends with low LCP content show higher rates as compared to PTT. High K g values for the other compositions is indicative of the larger barrier faced by them.

L–H plots for pure PTT using a t 0.1, b t 0.5. The other figures represent L–H plots for c 90/10, d 80/20, e 70/30, f 60/40 compositions using t 0.1 values

Variation of 1/t 0.1 as a function of undercooling
The regime transition temperature (Table 4) changes from 188 °C for PTT to 190 °C for the blends and once again shifts to 188 °C for the 60/40 composition. The regime transition for PTT is lower than the values 194–195 °C obtained by others [55, 60, 61]. Chen et al. [62] found that the regime transition temperature varied from 192 to 205 °C, decreasing with an increase in molecular weight. The current low value may be the result of the sample undergoing different thermal history.
The shift in the transition temperature to higher temperatures can be explained on the basis of i and g. As stated previously, the regime behaviour is determined by the competitive process between surface nucleation i and substrate completion rate g. The substrate completion rate is thought to have greater dependence on the reptation ability and thus the mobility of the polymer chains [63–65]. The slight increase in the T g observed for the blends is indicative of the constraints placed on the mobility of the chains. This can affect the process of zippering down into the niche created by the deposited stem, thus decreasing the lateral spread rate g [63]. More importantly, due to lowered energy barrier, the minor phase may also promote multiple nucleation events on the growth surface [8]. Such an increase in i coupled with decrease in g can cause a shift in the regime II to III transition temperature to higher temperatures. For the blends with high LCP content, a diminished rate of nucleation would cause regime transition temperature to shift to the lower temperature side.
The lateral surface energy can be estimated from the Thomas–Stavely relation [34, 66]:
where a is the molecular chain width taken to be 4.64 Å [55] and α was taken to be 0.18 [61, 55]. For PTT, b is taken as the perpendicular distance between (010) planes (5.71 Å) [55] and ΔH f has a value of 2.1 × 108 Jm−3 [55]. Thus, using the above relation σ was calculated to be 19.5 mJ m−2.
Using the above result, the fold surface energy was determined from the σσ e data listed in Table 4. The obtained values can be used to determine the work of chain folding (q), i.e. the work carried out in bending a polymer chain onto itself, using the following relation [37]:
where ab is the cross-sectional area. The values of σ e and q are listed in Table 5. The value of q obtained for regime II for PTT, i.e. 5.68 kcal mol−1, is close to 6.2 kcal mol−1 obtained by Hong et al. [61]. The blends show lower σ e values, especially at low LCP content. As stated previously, the inclusion of the minor phase may promote an increase in surface nucleation rate. This can result in the formation of a higher degree of nonadjacent loose folds and can contribute to the overall reduction in the fold surface energy. q, which depends on σ e, shows a similar decrease for blends with low LCP content indicating an easing of the chain folding process. Higher σ e values at other compositions indicate a decrease in the entropy of folding and thus the formation of more regular folding [36].
Conclusions
PTT/LCP blends were found to form primarily an immiscible system. During cooling, the LCP-rich phase crystallizes earlier and nucleates the PTT crystals, which subsequently reject the noncrystallizable material in the interfibrillar region, especially for low LCP concentrations. For blends with high LCP content, it is rejected into the interspherulitic region. The nucleating ability was seen through the increase in the overall crystallization rate represented by half-time of crystallization and Avrami rate constant. The k values increased with LCP content up to 70/30 composition decreasing slightly for the 60/40 compositions, but remaining above PTT. The studies on the melting behaviour revealed that the addition of LCP at low concentrations resulted in the formation of imperfect primary crystals. The Lauritzen–Hoffman analysis revealed that the blends with low LCP content show lower free energy barrier as well as low fold surface energies.
References
Siegmann A, Dagan A, Kenig S. Polyblends containing a liquid crystalline polymer. Polymer. 1985;26(9):1325–30.
Isayev AI, Modic M. Self-Reinforced melt processible polymer composites: extrusion, compression, and injection molding. Polym Compos. 1987;8(3):158–75.
Kiss G. In situ composites: blends of isotropic polymers and thermotropic liquid crystalline polymers. Polym Eng Sci. 1987;27(6):410–23.
Weiss RA, Huh W, Nicolais L. Novel reinforced polymers based on blends of polystyrene and a thermotropic liquid crystalline polymer. Polym Eng Sci. 1987;27(9):684–91.
Kalkar AK, Deshpande AA, Kulkarni MJ. In situ composites from blends of polycarbonate and a thermotropic liquid-crystalline polymer: the influence of the processing temperature on the rheology, morphology, and mechanical properties of injection-molded microcomposites. J Appl Polym Sci. 2007;106(1):34–45.
Tjong SC. Structure, morphology, mechanical and thermal characteristics of the in situ composites based on liquid crystalline polymers and thermoplastics. Mater Sci Eng R Rep. 2003;41(1–2):1–60.
Kalkar AK, Deshpande AA. Kinetics of isothermal and non-isothermal crystallization of poly(butylene terephthalate)/liquid crystalline polymer blends. Polym Eng Sci. 2001;41(9):1597–615.
Kalkar AK, Deshpande VD, Kulkarni MJ. Isothermal crystallization kinetics of poly(phenylene sulfide)/TLCP composites. Polym Eng Sci. 2009;49(2):397–417.
Yan H, Xu J, Mai K, Zeng H. Crystallization of poly(butylene terephthalate) blends containing liquid crystalline polymer component. Polymer. 1999;40(17):4865–75.
Minkova LI, Paci M, Pracella M, Magagnini P. Crystallization behavior of polyphenylene sulfide in blends with a liquid crystalline polymer. Polym Eng Sci. 1992;32(1):57–64.
Tjong SC, Chen SX, Li RKY. Crystallization kinetics of compatibilized blends of a liquid crystalline polymer with polypropylene. J Appl Polym Sci. 1997;64(4):707–15.
Minkova L, Magagnini PL. Blends of poly(ethylene 2,6-naphthalate) with liquid-crystalline polymers: crystallization behavior and morphology. Polymer. 2001;42(13):5607–13.
Bhattacharya SK, Tendolkar A, Misra A. Blends of a liquid crystalline polyester with polyethylene terephthalate. Mol Cryst Liq Cryst Incorp Nonlinear Opt. 1987;153(1):501–13.
de Carvalho B, Bretas RES. Crystallization kinetics of a PEEK/LCP blend. J Appl Polym Sci. 1995;55(2):233–46.
Naffakh M, Gómez MA, Ellis G, Marco C. Thermal properties, structure and morphology of PEEK/thermotropic liquid crystalline polymer blends. Polym Int. 2003;52(12):1876–86.
Naffakh M, Gómez MA, Ellis G, Marco C. Isothermal crystallization kinetics of PEEK/Vectra® blends by DSC and time-resolved synchrotron X-ray diffraction. Polym Eng Sci. 2006;46(10):1411–8.
Minkova LI, Magagnini PL. Non-isothermal crystallization kinetics of poly(phenylene sulfide)/Vectra-B blends. Polymer. 1995;36(10):2059–63.
Gao X, Liu R, Zhang J, Zhang J. Crystallization behaviors of poly(ethylene 2,6-naphthalate) in the presence of liquid crystalline polymer. Ind Eng Chem Res. 2008;47(8):2590–6.
Gabellini G. Bretas RES. Poly(p-phenylene sulfide)/liquid crystalline polymer blends. II. Crystallization kinetics. J Appl Polym Sci. 1996;61(10):1803–12.
Gopakumar TG, Ghadage RS, Ponrathnam S, Rajan CR, Fradet A. Poly(phenylene sulfide)/liquid crystalline polymer blends: 1. Non-isothermal crystallization kinetics. Polymer. 1997;38(9):2209–14.
Marinelli AL, Bretas RES. Blends of polypropylene resins with a liquid crystalline polymer. I. Isothermal crystallization. J Appl Polym Sci. 2003;87(6):916–30.
Georgiev G, Cebe P, Capel M. Effects of nematic polymer liquid crystal on crystallization and structure of PET/Vectra blends. J Mater Sci. 2005;40(5):1141–52.
Budgell DR, Day M. Crystallization behavior of polyphenylenesulfide. Polym Eng Sci. 1991;31(17):1271–8.
Saito H, Okada T, Hamane T, Inoue T. Crystallization kinetics in the mixtures of poly(vinylidene fluoride) and poly(methyl methacrylate): two-step diffusion mechanism. Macromolecules. 1991;24(15):4446–9.
Lee S, Mather PT, Pearson DS. Phase behavior and rheology of blends containing polycarbonate and a thermotropic polyester. J Appl Polym Sci. 1996;59(2):243–50.
Tjong SC, Li RKY, Xie X. Properties of in situ composites based on semiflexible thermotropic liquid crystalline copolyesteramide and polyamide 66 blends. Polym J. 2000;32(11):907–14.
Xie XL, Li RKY, Tjong SC, Tang CY. Flory–huggins interaction parameters of LCP/thermoplastic blends measured by DSC analysis. J Therm Anal Calorim. 2002;70(2):541–8.
Zhong Y, Xu J, Zeng H. Blends of poly(ether ether ketone) with a thermotropic liquid crystalline polyester I. The morphology, crystallization, and melting behavior. Polym J. 1992;24(10):999–1007.
Pisitsak P, Magaraphan R. Non-isothermal crystallization kinetics and melting behaviors of thermoplastic/liquid crystalline polymer blends of poly(trimethylene terephthalate)/Vectra A950. Adv Sci Technol. 2008;54:249–54.
Pisitsak P, Magaraphan R. Influences of a liquid crystalline polymer, vectra A950, on crystallization kinetics and thermal stability of poly(trimethylene terephthalate). J Therm Anal Calorim. 2009;95(2):661–6.
Avrami M. Kinetics of phase change. I General theory. J Chem Phys. 1939;7(12):1103–12.
Avrami M. Kinetics of phase change. II Transformation-time relations for random distribution of nuclei. J Chem Phys. 1940;8(2):212–24.
Avrami M. Granulation, phase change, and microstructure kinetics of phase change. III. J Chem Phys. 1941;9(2):177–84.
Hoffman JD, Davis GT, Lauritzen JI. The rate of crystallization of linear polymers with chain folding. In: Hannay NB, editor. Treatise on solid state chemistry. New York: Plenum Press; 1976. p. 497–605.
Phetwarotai W, Aht-Ong D. Isothermal crystallization behaviors and kinetics of nucleated polylactide/poly(butylene adipate-co-terephthalate) blend films with talc. J Therm Anal Calorim. 2016;126(3):1797–808.
Di Lorenzo ML. Spherulite growth rates in binary polymer blends. Prog Polym Sci. 2003;28(4):663–89.
Hoffman JD, Miller RL. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: theory and experiment. Polymer. 1997;38(13):3151–212.
Hoffman JD, Weeks JJ. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J Res NatL Bur Stand A. 1962;66A(1):13–28.
Chung T-S, Cheng M, Goh SH, Jaffe M, Calundann GW. Revisit the crystallization mechanism of vectra,™ a liquid crystal polymer. J Appl Polym Sci. 1999;72(9):1139–50.
Liang H, Xie F, Wu W, Guo F, Chen B, Xu J. Miscibility and melting properties of poly(ethylene 2,6-naphthalate)/poly(trimethylene terephthalate) blends. J Mater Sci. 2008;43(8):2739–44.
Groeninckx G, Vanneste M, Everaert V. Crystallization, morphological structure, and melting of polymer blends. In: Utracki LA, editor. Polymer blends handbook. Dordrecht: Kluwer Academic Pub; 2002. p. 203–89.
Lorenzo AT, Arnal ML, Albuerne J, Müller AJ. DSC isothermal polymer crystallization kinetics measurements and the use of the Avrami equation to fit the data: guidelines to avoid common problems. Polym Test. 2007;26(2):222–31.
Long Y, Shanks RA, Stachurski ZH. Kinetics of polymer crystallisation. Prog Polym Sci. 1995;20(4):651–701.
Chisholm BJ, Zimmer JG. Isothermal crystallization kinetics of commercially important polyalkylene terephthalates. J Appl Polym Sci. 2000;76(8):1296–307.
Binsbergen FL. Natural and artificial heterogeneous nucleation in polymer crystallization. J Polym Sci Polym Symp. 1977;59(1):11–29.
Kalkar AK, Deshpande VD, Kulkarni MJ. Nonisothermal crystallization kinetics of poly(phenylene sulphide) in composites with a liquid crystalline polymer. J Polym Sci Part B Polym Phys. 2010;48(10):1070–100.
Ho R-M, Ke K-Z, Chen M. Crystal structure and banded spherulite of poly(trimethylene terephthalate). Macromolecules. 2000;33(20):7529–37.
Shafee EE, Naguib HF, Li L, Jiang S, An L. Crystallization and spherulitic growth kinetics of poly(trimethylene terephthalate)/polycarbonate blends. Polym Eng Sci. 2010;50(5):1036–46.
Bassett DC. Principles of polymer morphology. Cambridge: Cambridge University Press; 1981.
Kim GS, Son JM, Lee JK, Lee KH. Morphology development and crystallization behavior of poly(ethylene terephthalate)/poly(trimethylene terephthalate) blends. Eur Polym J. 2010;46(8):1696–704.
Keith HD, Padden FJ. Spherulitic crystallization from the Melt. I. Fractionation and impurity segregation and their influence on crystalline morphology. J Appl Phys. 1964;35(4):1270–85.
Keith HD, Padden FJ. Spherulitic crystallization from the Melt. II. Influence of fractionation and impurity segregation on the kinetics of crystallization. J Appl Phys. 1964;35(4):1286–96.
He Y, Zhu B, Kai W, Inoue Y. Effects of crystallization condition of poly(butylene succinate) component on the crystallization of poly(ethylene oxide) component in their miscible blends. Macromolecules. 2004;37(21):8050–6.
Srimoaon P, Dangseeyun N, Supaphol P. Multiple melting behavior in isothermally crystallized poly(trimethylene terephthalate). Eur Polym J. 2004;40(3):599–608.
Supaphol P, Apiwanthanakorn N, Krutphun P. Effect of small amount of poly(ethylene naphthalate) on isothermal crystallization and spherulitic morphology of poly(trimethylene terephthalate). Polym Test. 2007;26(8):985–1000.
Liang H, Xie F, Chen B, Guo F, Jin Z, Luo F. Miscibility and melting behavior of poly(ethylene terephthalate)/poly(trimethylene terephthalate) blends. J Appl Polym Sci. 2008;107(1):431–7.
Bulakh N, Jog JP. Crystallization of poly(phenylenesulfide)/amorphous polyamide blends: DSC and microscopic studies. J Macromol Sci Part B. 1999;38(3):277–87.
Chan TW, Isayev AI. Quiescent polymer crystallization: modelling and measurements. Polym Eng Sci. 1994;34(6):461–71.
Gupta A, Simmons W, Schueneman GT, Mintz EA. Lignin-coated cellulose nanocrystals as promising nucleating agent for poly(lactic acid). J Therm Anal Calorim. 2016;126(3):1243–51.
Achilias DS, Papageorgiou GZ, Karayannidis GP. Evaluation of the crystallisation kinetics of poly(propylene terephthalate) using DSC and polarized light microscopy. J Therm Anal Calorim. 2006;86(3):791–5.
Hong P-D, Chung W-T, Hsu C-F. Crystallization kinetics and morphology of poly(trimethylene terephthalate). Polymer. 2002;43(11):3335–43.
Chen C-C, Chen M, Tseng IM. the influence of molecular weight on the regime crystallization of poly(trimethylene terephthalate). J Macromol Sci Part B. 2002;41(4–6):1043–61.
Phillips PJ, Lambert WS. Regime transitions in a non-reptating polymer: crosslinked linear polyethylene. Macromolecules. 1990;23(7):2075–81.
Scott Lambert W, Phillips PJ. Crystallization kinetics of fractions of branched polyethylenes: 2. Effect of molecular weight. Polymer. 1996;37(16):3585–91.
Xu J-T, Zhao Y-Q, Wang Q, Fan Z-Q. Observation of regime III crystallization in polyethylene/montmorillonite nanocomposites. Macromol Rapid Commun. 2005;26(8):620–5.
Thomas DG, Staveley LAK. A study of the supercooling of drops of some molecular liquids. J Chem Soc. (Resumed). 1952:4569–77.
Acknowledgements
The authors would like to thank Mr. Kailash Dangayach of Shell and Professor Kanitkar of ICT for their help in obtaining the samples. The authors would also like to thank Prof. Kalkar for his inputs. Mr. Sandeep Jape has received fellowship from University Grants Commission, Government of India, New Delhi, under Special Assistance Program (SAP); contract grant number: F.4-1/2006(BSR)/5-65/2007. He has since then received financial assistance from TEQIP.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deshpande, V.D., Jape, S.P. Morphology, crystallization and melting behaviour of poly(trimethylene terephthalate)/thermotropic liquid crystalline polymer blends. J Therm Anal Calorim 128, 1479–1493 (2017). https://doi.org/10.1007/s10973-017-6089-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6089-7