
Abstract
A series of complexes [M2L2Cl2(OH2) n ]·mH2O (M: Co, n = 0, m = 1; M: Ni, n = 2, m = 0; M: Cu, n = 0, m = 0; M: Zn, n = 0, m = 1; HL·H2O: 2-[(E)-1H-1,2,4-triazol-3-ylimino)methyl]phenol) were synthesised and characterised. The features of complexes have been assigned from microanalytical, thermal, IR, UV–Vis-NIR, EPR spectroscopy as well as magnetic data at room temperature. The electrochemical behaviour of the complexes was also investigated by cyclic voltammetry. The simultaneous TG/DTA experiments of these in flowing air atmosphere evidenced the occurrence of water or chloride as well as oxidative degradation of the Schiff base. The final product of decomposition was the most stable metal oxide as powder X-ray diffraction indicates. The complexes exhibited an improved antibacterial activity in comparison with the ligand towards both planktonic as well as biofilm embedded cells. The Cu(II) and Zn(II) complexes exhibited an inhibitory effect on the tumor cell lines growth, as revealed by the increased G2 phase and percentage of apoptotic/necrotic cells.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The resistance occurrence after the introduction of antibiotic and cytostatic drugs in the clinical practice represents an emerging problem of current interest. Furthermore, the pathogenic micro-organisms have the ability to develop biofilms on prosthetic materials or natural tissues, feature which is regulated by an ubiquitary mechanism involved in the coordinated expression of virulence factors, including the adherence ability [1–4].
In order to solve these problems, many complexes were designed including those with multifunctional Schiff bases as ligands. Some of these studies were devoted to the synthesis and characterization of some derivatives bearing 1,2,4-triazole scaffold, studies generated by the fact that 1,2,4-triazoles possess several useful features for new drug design. The triazole ring is chemically stable to acidic and basic hydrolysis as well as to reductive and oxidative conditions, and it is relatively resistant to metabolic degradation [5, 6]. Moreover, the triazole derivatives have the ability to generate noncovalent interactions such as hydrogen bonding as well as dipole–dipole and π-stacking ones [7, 8]. Consequently, this ring can interact with biological targets and represents a powerful pharmacophore for several drugs that are already in use for the treatment or symptom amelioration of a wide range of diseases such microbial, viral and parasitic infections, malignant tumours, epilepsy, diabetes, obesity, cardiovascular, neurological, immunological as well as psychiatric disorders [8–12].
Recent studies evidenced a good anti-inflammatory [13, 14], antimicrobial [15, 16] and antitumour [15, 17] activity for some polyfunctional Schiff base derived from 1,2,4-triazole. As a result, 1,2,4-triazole-containing Schiff base complexes were also studied and some of that demonstrated ability to catalyse superoxide radical decomposition [18] or to inhibit protein tyrosine phosphatase [19]. The anticonvulsant [20], antitumour [21] or antimicrobial activity [22–28] was also evidenced for complexes with such ligands. In all cases, it was evidenced that overall antimicrobial potency of the uncoordinated multifunctional Schiff bases was enhanced upon coordination [22–28].
In the present work, we report the synthesis of Co(II), Ni(II) Cu(II) and Zn(II) complexes with Schiff base derived from 3-amino-4H-1,2,4-triazole and salicylaldehyde. The complexes were characterised by elemental analyses, IR and UV–Vis-NIR, EPR spectra as well as magnetic susceptibility and thermogravimetric analysis. The thermal behaviour of these derivatives was investigated in synthetic air by simultaneously TG/DTA measurements in order to evidence the complexes modifications at heating.
The complexes were designed as metal-based biologically active candidates by combining the chemistry of some essential ions with that of Schiff bases bearing 1,2,4-triazole moiety that could act against both genetically resistant as well as biofilm-embedded pathogenic strains. The compounds cytotoxicity was also evaluated on tumour cell lines Hep 2 and HCT 8, respectively.
Experimental
Materials
The high purity reagents were obtained commercially from Sigma-Aldrich (CoCl2·6H2O, NiCl2·6H2O, CuCl2·2H2O, ZnCl2), Loba (salicylaldehyde) and Fluka (3-amino-4H-1,2,4-triazole) and were used as received without further purification.
Instruments
Chemical analysis of carbon, nitrogen and hydrogen has been performed using a Perkin Elmer PE 2400 analyser.
Metal content was determined by atomic absorption spectrophotometry (AAS) on a Avanta GBC spectrometer by using a stock standard solution (Merck,1 g mL−1), while the working solutions were prepared by a suitable dilution of the sample obtained by complex calcination at 450 °C and the successive treatment of the residue with HCl and HNO3.
IR spectra were recorded in KBr pellets with a Bruker Tensor 37 spectrometer in the range of 400–4,000 cm−1.
1H NMR spectra were recorded on a Bruker DPX200 spectrometer (working frequency 200 MHz) at 25 °C. Chemical shifts were measured in parts per million from internal standard TMS.
Electronic spectra by diffuse reflectance technique, with spectralon as standard, were recorded in the range of 200–1,500 nm, on a Jasco V670 spectrophotometer.
Magnetic measurements were done at room temperature, on a Lake Shore’s fully integrated vibrating sample magnetometer (VSM) system 7,404, calibrated with a Ni—0.126 g sphere—SRM 772a. The VSM was intercalibrated either with an absolute calibrated Faraday-balance or with Hg[Co(NCS)4] as standard. The molar magnetic susceptibilities were calculated and corrected for the atomic diamagnetism.
The X-band EPR measurements were recorded at 293 K for solid sample and at 100 K for DMSO solution on a JEOL FA100 spectrometer. The general settings used were as follows: sweep field 1,000 G, frequency 100 kHz, gain in the range of 100–200, sweep time 1,800 s, time constant 1 s, modulation width 2 G, microwave power 1 mW. The magnetic field calibration was performed with a DPPH (diphenylpicrylhydrazyl) standard marker, exhibiting a narrow EPR line at g = 2.0036.
Cyclic voltammograms were recorded by an electrochemical system (potentiostate/galvanostate) Autolab PGSTAT 12. Electrochemical studies were performed at room temperature under inert atmosphere (Ar 99.9999 %) in DMSO-containing tetrabutylammonium perchlorate (Bu4NClO4) 0.1 M as supporting electrolyte. The reference electrode was Ag/AgCl (LiCl saturated in ethanol). The counter electrode was the platinum wire. The working electrode was a glassy carbon (GC) with the effective aria of electrode 7.065 mm2. The details concerning electrochemical behaviour of the compounds are presented as Online Resources 1.
The heating curves (TG, DTG and DTA) were recorded using a simultaneous TG/DTA Labsys 1200 SETARAM instrument, with a sample mass of 9–15 mg over the temperature range of 20–900 °C with a heating rate of 10 °C min−1. The measurements were carried out in synthetic air atmosphere (flow rate 17 cm3 min−1) by using alumina crucibles. The calibration of TG equipment was run both with gold (m. p. 1,064 ± 1 °C) and ultrapure CuSO4·5H2O (99.9 %) with a measurement error of 0.1 %.
The X-ray powder diffraction patterns were collected on a PANalytical X’Pert PRO diffractometer using Cu K α radiation (λ = 1.5406 Å) in the range of 2θ from 5° to 80°.
Antimicrobial assays
The antimicrobial activities of the complexes were determined against reference and clinical isolates Gram-negative (Escherichia coli ATCC 25922, E. coli 832, Klebsiella pneumoniae ATCC 134202, K. pneumoniae 806, Pseudomonas aeruginosa ATCC 27853) and Gram-positive (Staphylococcus aureus ATCC 25923, Bacillus subtilis ATCC 6633) bacterial strains, as well as Candida albicans 22 fungal strain. Microbial suspensions of 1.5 × 108 CFU mL−1 (0.5 McFarland density) obtained from 15 to 18 h bacterial cultures developed on solid media were used. The antimicrobial activity was tested on gelose medium. The compounds (ligand and complexes) were solubilised in DMSO and the starting stock solution was of 1,000 μg mL−1 concentration. The qualitative screening was performed by an adapted disc diffusion method as was previously reported [29].
The quantitative assay of the antimicrobial activity was performed by the liquid medium microdilution method, in 96-multi-well plates, in order to establish the minimal inhibitory concentration (MIC). In this purpose, serial two-fold dilutions of the compounds ranging between 1,000 and 1.95 µg mL−1 were performed in a 200 µL volume of broth and each well was seeded with 20-μL microbial inoculum. Sterility control (wells containing only culture medium) and culture controls (wells containing culture medium seeded with the microbial inoculum) were used. The influence of the DMSO solvent was also quantified in a series of wells containing DMSO, diluted accordingly with the dilution scheme used for the complexes. The plates were incubated for 24 h at 37 °C, and MIC values were considered as the lowest concentration of the tested compound that inhibited the visible growth of the microbial overnight cultures [30].
The assessment of the complexes influence on the microbial ability to colonize an inert substratum was performed by the micro-titre method, following previously described protocols [31]. The absorbance at 490 nm was measured with an ELISA reader Apollo LB 911. All biological experiments were performed in triplicates.
Cell viability assay and apoptosis
Hep 2 (CCL 223™) and HCT 8 cell lines (CCL 244™) were cultivated in RPMI 1640 (Gibco, NY, SUA) supplemented with 10 % heat-inactivated bovine serum and penicillin/streptomycin at 37 °C with 5 % CO2.
To prepare cells for plating into 24-well plate, adherent cells were detached, centrifuged, suspended in fresh medium, counted by trypan blue exclusion and adjusted to 5 × 104 cells mL−1. Cells were left for 24 h at 37 °C, in 5 % CO2 prior to addition of newly synthesised substances. The substances were initially dissolved into DMSO, and added to the cell culture in a final concentration of 100 µg mL−1.
In order to discriminate between intact and treatment affected cells, we used Annexin V-FITC Apoptosis Detection Kit I (BD Bioscience Pharmingen, USA), according to manufacturer protocol. Briefly, the total cells were suspended in 100 µL of binding buffer (10 mM of HEPES/NaOH, pH 7.4, 140 mM NaCl and 2.5 mM CaCl2), and stained with 5 µL Annexin V-FITC and 5 µL propidium iodide (PI) for 10 min in dark prior to analyse using a Beckman Coulter flow cytometer. At least 10,000 events from each sample were acquired. The percentage of treatment-affected cells was determined using FlowJo software [32, 33].
Cell cycle distribution
A quantity of 105 HCT 8 cells/well were plated in 24-well plate and treated for 24 h with 100 µg mL−1 compound solution. After treatment period, cells were taken from the substrate, fixed in 70 % cold ethanol for at least 30 min at −20 °C, washed twice in phosphate-buffered saline (PBS) and then incubated for 15 min at 37 °C with RNAse A (100 µg mL−1) and 1 h with propidium iodide (100 µg mL−1). After this treatment, the data acquisition was done using Epics Beckman Coulter flow cytometer. Data were analysed using FlowJo software and expressed as the fractions of cells in the different cell cycle phases [32, 33].
Synthesis, analytical and spectral data for ligand and complexes
To a solution containing 5 mmol cobalt(II) chloride hexahydrate (1.19 g) in 25 mL−1 ethanol was added 5 mmol salicylaldehyde (0.61 g, ρ = 1.146 g cm−3) and a solution containing 5 mmol 3-amino-4H-1,2,4-triazole (0.42 g) in 25 mL−1 ethanol. The reaction mixture was magnetically stirred at 50 °C for 4 h, until a sparingly soluble species was formed. The precipitate was filtered off, washed several times with cold ethanol and air-dried.
The same method was used for the preparation of all complexes from the series. The analytical data for these species are given below.
[Co2L2Cl2]·H2O (1): Yield: 63 %; Analysis, found (%): Co, 20.12; C, 37.04; H, 2.65; N, 19.34; Co2C18H16N8O3Cl2 requires (%): Co, 20.28; C, 37.20; H, 2.78; N, 19.28.
[Ni2L2Cl2(OH2)2] (2): Yield: 94 %; Analysis, found (%): Ni, 19.03; C, 36.06; H, 3.17; N, 18.97; Ni2C18H18N8O4Cl2 requires (%): Ni, 18.93; C, 36.11; H, 3.03; N, 18.72.
[Cu2L2Cl2] (3): Yield: 70 %; Analysis, found (%): Cu, 22.04; C, 37.57; H, 2.31; N, 19.71; Cu2C18H14N8O2Cl2 requires (%): Cu, 22.21; C, 37.77; H, 2.47; N, 19.58.
[Zn2L2Cl2]·H2O (4): Yield: 58 %; Analysis, found (%): Zn, 21.87; C, 36.26; H, 2.58; N, 18.98; Zn2C18H16N8O3Cl2 requires (%): Zn, 22.02; C, 36.39; H, 2.71; N, 18.86.
The ligand was synthesised by a literature procedure [34] and identified through chemical analysis, IR (Table 1) and 1H NMR spectra. 1H NMR (DMSO) δ (ppm): 5.77 (s, NH), 7.04 (dd, J = 6.40, 3.00 Hz, Ar–H), 7.49 (dd, J = 6.78; 2.50 Hz, Ar–H), 7.81 (d, J = 6.10 Hz, Ar–H), 8.46 (s, CH = N), 9.46 (s, CH), 12.40 (s, OH).
Results and discussions
Synthesis, spectral and magnetic data for complexes
The ligand was synthesised by [1 + 1] condensation of 3-amino-4H-1,2,4-triazole and salicylaldehyde by a method reported in the literature [34]. The [M2L2Cl2(OH2) n ]·mH2O (M: Co, n = 0, m = 1; M: Ni, n = 2, m = 0; M: Cu, n = 0, m = 0; M: Zn, n = 0, m = 1; HL: 2-[(E)-1H-1,2,4-triazol-3-ylimino)methyl]phenol) were obtained either by direct reaction of Schiff base and metal chlorides in a 1:1 molar ratio or by template process conducted in a 1:1:1 molar ratio, as presented in Scheme 1. The chemical and thermal analyses indicate the self deprotonation of Schiff base upon metallic ion coordination.

Synthetic route to prepare complexes and coordination proposed
Infrared spectra
The most relevant IR bands and assignments for the ligand and complexes are summarised in Table 1. The IR spectra of all complexes display bands at 3,123–3,128, 1,542–1,606 and 1,055–1,078 cm−1 that originate from triazole precursor [24, 27, 28]. The bands located in 3062–3088, 1463–1468 and 761–768 cm−1 ranges, respectively, represent a pattern for ortho-substituted benzene ring. In the IR spectra of the Schiff base an intense band at 1,615 cm−1 that is missing in the 3-amino-1,2,4-triazole spectrum, provides a strong evidence for condensation and azomethine group formation. This band is shifted towards higher wavenumbers by 23–49 cm−1 in complexes spectra indicating the involvement of azomethine nitrogen atom in coordination [22, 35]. In the same range at 1,575 cm−1 appears a very intense band in the ligand spectrum assigned to ν(C=N) from triazole ring. For Ni(II) complex in this range, two bands can be observed, one being shifted to lower wavenumbers, feature considered an indicative of one nitrogen atom from triazole ring coordination [24, 27]. The band at 1,277 cm−1 assigned to ν(C–O) vibration mode is shifted also towards lower wavenumbers by 40–60 cm−1 in comparison with ligand as result of both deprotonation and phenolic oxygen coordination [36, 37]. In the characteristic ranges for water, a broad band around 3,420 cm−1 can be assigned to ν(OH) stretching vibration, except for complex (2). Additional bands in the range of 660–770 cm−1 indicate that some water molecules are coordinated in the case of complex (2) [38]. In the ranges 430–470 and 470–520 cm−1, two supplementary bands, tentatively assigned to stretching vibrations ν(M–N) and ν(M–O), appear in the spectra of complexes [38]. These bands represent a proof for Schiff base coordination through azomethine nitrogen and phenolic oxygen.
1H NMR spectrum
The 1H NMR spectrum of ligand exhibit a multiplet in the range of 7.04–7.81 ppm assigned to phenyl protons. The proton of-OH group at 2-position of benzene ring appears as a singlet at 12.40 ppm, while the signal at 8.46 ppm is characteristic for azomethine group proton [39].
The 1H NMR spectrum of complex (4) was not possible to register having in view the low solubility of this compound in all deuterated solvents.
Electronic spectra and magnetic moments
Electronic spectra combined with magnetic moments at room temperature could provide useful information concerning the oxidation state of the metallic ion, stereochemistry as well as the ligand field strength. Table 2 lists the electronic absorption bands, their assignments as well the magnetic moments at room temperature.
Two intense bands can be noticed in the ligand spectrum assigned to π → π* transition arising from both C=N groups of azomethine and triazole moieties. The first one is shifted in the complexes spectra as result of deprotonation and/or coordination, while the second one is obscured by the complexes bands that appear in the same region.
Five additionally bands appear in the electronic spectrum of the cobalt(II) complex assigned to spin allowed transitions 4 A 2 → 4 T 1(F) and 4 A 2 → 4 T 1(P) for tetrahedral geometry. These components appear as result of the split of the two usually observed for this ion in the visible and NIR regions due to distortion generated by a different nature of donor atoms and ligands, respectively [40]. The Co(II) complex exhibits a magnetic moment of 4.36 B.M. at room temperature, value that lies in the range accepted for tetrahedral species (4.2–4.8 B.M.) with three unpaired electrons and a very low orbital contribution [41].
The Ni(II) complex (2) exhibits an octahedral geometry as indicate the three bands that can be assigned to 3 A 2g → 3 T 2g, 3 A 2g → 3 T 1g(F) and 3 A 2g → 3 T 1g(P) spin allowed transitions, respectively [40]. The low intensity band at 12,660 cm−1 is assigned to a spin forbidden transition. A value of 0.91 for nephelauxetic parameter indicates a low degree of covalency. The room temperature magnetic moment of 2.90 B.M., close to the spin-only magnetic moments value (2.83 B.M.) for two unpaired electrons, further supports this stereochemistry [41].
The electronic spectrum of the Cu(II) complex (Fig. 1) exhibits a band with a maximum at 12,990 cm−1 tentatively assigned to \( d_{xy}\rightarrow d_{{x}^{2}-{y}^{2}}\) transition in a square planar stereochemistry and a weak field associated with chlorine and oxygen donor atoms. The value of 1.49 B.M. for the magnetic moment is characteristic for Cu(II) compounds with interaction between paramagnetic ions (S = ½) at room temperature [41].

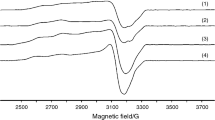
EPR spectra at room temperature in powder (a), in DMSO (b) and electronic spectrum of complex (3) (onset)
In order to minimize the repulsions, the tetrahedral stereochemistry was tentatively proposed for Zn(II) complex considering the volume of chloride anion and the negatively charge located on both ligands [41].
EPR spectra
The powder EPR spectrum for (3) exhibits a broad line with an approximately 380 G width suggesting a strong interaction between paramagnetic centres (Fig. 1) [42]. This aspect of the spectrum can be related to a square planar stereochemistry with grossly misaligned tetragonal axes. The EPR spectrum of (3) in DMSO-frozen solution shows an asymmetric signal indicating an octahedral stereochemistry with a rhombic distortion and the g values: g 1 = 2.225, g 2 = 2.147 g 3 = 2.111 and a hyperfine splitting constant A ll (≈200 G). This behaviour can arise from DMSO coordination in one or both axial vacant coordination sites.
Electrochemistry
Cyclic voltammograms of both solvated Co(II) and complex (1) (Fig. S1) have similar shapes. The cathodic peak, corresponding to the Co(II)/Co(I) reduction [43], is shifted towards more negative potentials and its intensity is reduced by an order of magnitude for complex (1) in comparison with that observed for the solvated Co(II) (Table S1), indicating that the Schiff base coordination renders the reduction process more difficult. The Co(II)/Co(III) oxidation process is also hampered by organic ligand complexation as it can be observed by both anodic peak potential shifting to positive value and by peak current intensity that decreased dramatically.
In the case of the complex (2), a similar behaviour was observed for the cathodic peak assigned to Ni(II) reduction and the peak due to Ni(II)/Ni(III) oxidation [43] (Table S1).
In the cyclic voltammograms of Cu(II) species (Fig. S1), the second cathodic peaks with E p2c = −0.68 V for the solvated Cu(II) ion and E p2c′ = −1.00 V for the complex (3), respectively, are due to Cu(II)/Cu(I) reduction. The solvated Cu(I) ion is unstable and disproportionate to Cu(II) and Cu(0), the last one being reoxidized in the reverse scan, giving rise to the anodic peaks observed between −0.20 and −0.10 V [43].
Complex (4) exhibits only a less intensive irreversible reduction peak compared to the quasireversible behaviour of the solvated Zn(II) species (Table S1).
Thermal behaviour
Thermal analysis techniques are frequently used in order to obtain useful informations concerning the composition and stability of complexes [44–46]. Moreover, the understanding of the thermal behaviour of complexes with biological activity represents an important step in order to develop further applications [47, 48]. In order to obtain such information, the thermal behaviour of complexes was investigated by simultaneous TG/DTA analysis, while the final residue was examined by powder X-ray diffraction. The species isolated after the water elimination was also isolated and characterised in order to establish its presence as ligand or as hydration species.
The TG, DTG and DTA curves corresponding to the complex (1) indicate that decomposition follows three steps (Fig. 2). The first step of compound thermal transformation consists in an endothermic release of water molecules up to 145 °C (Table 3). The low temperature interval that corresponds with this elimination indicates the crystallisation nature of these molecules [49]. This role of water molecule is furthermore supported by the same pattern of the electronic spectra of complex and its residue at 140 °C. The anhydrous species then suffers endothermic chloride elimination, the complex neutrality being assured by the oxo anion. It is to be pointed the negative reaction for the chloride anions with silver nitrate for the residue isolated at 400 °C. Such behaviour was observed for other chloride complexes during the thermal decomposition [50]. The third step is not a single one but an overlap of several processes according to both DTG and DTA curves profile. According to the mass loss in the last step, Co(II) into Co(III) transformation together with oxidative degradation of Schiff base occurs. All these thermal transformations lead to Co3O4 as final product (found/calcd. overall mass loss: 72.1/72.4 %) (ASTM 78-1970). The endothermic effect noticed on DTA curve after the mass stabilisation can be assigned to Co3O4 crystallisation process.

TG, DTG and DTA curves for [Co2L2Cl2]·H2O (1)
For complex (2), the decomposition proceeds also in three steps (Fig. 3). In first endothermic step, coordination water is eliminated up to 170 °C as was observed for other similar complexes [51]. This assumption is furthermore supported by the electronic spectrum of intermediate isolated at about 170 °C that preserves the pattern of octahedral stereochemistry but the absorption maxima are different. Thus, the d–d transitions at 9,130, 15,870 and 25,315 cm−1 indicate an increase of crystalline field strength for intermediate, behaviour that can arise from the replacement of coordinated water molecules through nitrogen atoms from the triazole moiety. The chloride anions are endothermic released in the second step. The mass loss up to 575 °C corresponds to oxidative degradation of the Schiff base, and according to both DTA and DTG curves at least two overlapped processes occur until the nonstoichiometric NiO is formed (ASTM 78-0429) (found/calcd. overall mass loss: 74.7/75.0 %). This species loses continuously oxygen from the lattice considering that the TG curve slightly decreases and an exothermic effect can be noticed on DTA curve at temperatures over 600 °C.

TG, DTG and DTA curves for [Ni2L2Cl2(OH2)2] (2)
Complex (3) is an anhydrous species and as result their thermal degradation starts at 240 °C. On the other hand, the decomposition pattern is different from that of other species from the series (Fig. 4). First step is endothermic one and according to the mass loss corresponds to chloride anions elimination. Towards the end of the chloride replacement with oxo anion, the sample melts at 275 °C, process accompanied by a sharp endothermic effect on DTA curve. The Schiff base thermal decomposition starts immediately with 58 % from its mass elimination process accompanied by several exothermic overlapped processes according to DTG curve profile. The rest of organic matrix is oxidative eliminated in the last step with CuO formation according to powder X-ray diffraction (ASTM 5-661) (found/calcd. overall mass loss: 72.1/72.2 %). A slight mass loss accompanied by an exothermic effect can be noticed over 860 °C that can indicate a slow release of oxygen from lattice with generation of a nonstoichiometric species.

TG, DTG and DTA curves for [Cu2L2Cl2] (3)
The first step in the curve of compound (4) (Fig. 5) occurs in the temperature range of 90–145 °C, when the water molecules are loss in a well definite, endothermic step. The temperature range corresponding to this transformation indicates the crystallisation nature of these molecules [49]. The next step, endothermic also, corresponds to chloride displacement by oxo anion from the anhydrous complex. The third step, an exothermic and complex one is an overlap of at least four processes (as DTA curve profile indicates), and corresponds with the organic component pyrolysis and zinc(II) oxide generations, respectively (ASTM 036-1451) (found/calcd. overall mass loss: 72.5/72.6 %).

TG, DTG and DTA curves for [Zn2L2Cl2]·H2O (4)
Having in view all these data, a dimeric structure was proposed for all complexes in order to achieve the tetrahedral or octahedral stereochemistry and to correlate the IR and thermal data as is shown in Scheme 1.
Antimicrobial activity assay
The antimicrobial activity of the compounds was assayed against Gram-negative (E. coli ATCC 25922, E. coli 832, K. pneumoniae ATCC 134202, K. pneumoniae 806, P. aeruginosa ATCC 27853) and Gram-positive (S. aureus ATCC 25923, B. subtilis ATCC 6633) pathogenic bacterial strains, as well as against C. albicans 22 fungal strain. It is to be pointed that E. coli 832 and K. pneumoniae 806 strains were isolated from urine culture and wound secretion, respectively, exhibiting a multidrug resistance phenotype. It was observed that the DMSO solvent does not influence the antimicrobial activity of the tested compounds at the working concentrations. The assays revealed a very good antimicrobial activity for the complex (3) with a MIC value of 3.9 μg mL−1 in the case of E. coli ATCC 25922, 31.25 μg mL−1 against C. albicans 22 and 62.5 μg mL−1 for K. pneumoniae ATCC 134202. The same MIC value of 62.5 μg mL−1 was obtained for the complex (2) against C. albicans 22. The complex (4) exhibited a good activity with a MIC value of 31.25 μg mL−1 for B. subtilis ATCC 6633 (Table 4).
Overall, all the complexes exhibited an improved activity in comparison with the free ligand. This may arise from their ability as neutral species to cross the membrane by passive diffusion, and to accumulate in active concentration near by the intracellular microbial target. Thereby, the compounds lipophilicity and/or their ability to form coordinative or non-covalent bonds with the enzymes or other cytosolic components may result in the interference with the normal cell growth of pathogenic microorganisms. The most active compounds were (3) and (4) most probably due to their stereochemical versatility.
Having in view the differences in the physiology and susceptibility to antibiotics of biofilm-embedded microorganisms, the obtained complexes were investigated concerning their efficiency against the adherent cells grown in biofilms developed in plastic wells. From this point of view, it was observed that the complexes interact differently with the biofilm formed by different microbial strains, the effect being either inhibitory or stimulatory depending on the tested strain and the compounds concentration. The complexes (1) and (4) exhibited the most evident inhibitory effect upon de adherence ability of S. aureus ATCC 25923, B. subtilis ATCC 6633 and P. aeruginosa ATCC 27853, up to the concentration of 1.95 μg mL−1. The complex (3) exhibited an important inhibitory effect upon the adherence ability of the E. coli ATCC 25922 strain, up to the concentration of 3.9 μg mL−1 and of the C. albicans 22 fungal strain with a minimal biofilm eradication concentration (MBEC) of 31.25 μg mL−1. The anti-biofilm activity was expressed above the concentration of 7.81 μg mL−1 by complex (1) on C. albicans 22, (2) on P. aeruginosa ATCC 27853 and (4) on E. coli ATCC 25922 strain (Table S2).
Cytotoxicity assay
The cytotoxicity evaluation of the ligand and the obtained complexes was performed on two human tumour cell lines, i.e. Hep 2 (human cervix carcinoma HeLa derivative) and HCT 8 (human ileocecal adenocarcinoma), respectively.
The cell morphology observed after Hep 2 line treatment with 100 μg mL−1 of the obtained compounds indicates the fact that the most affected were cells we those treated with the compound (4), and less affected were those treated with compound (1). The cell viability decreased to 67.80 % for (3) and, respectively, to 45.00 % for (4) (Table S3).
In order to define the molecular events associated with cytotoxicity, the cellular responses induced by compounds were also studied by using the flow cytometry method. Thereby, it was observed that 100 μg mL−1 of compounds (3) and (4) induced the increase of G2-phase of HCT 8 cells after 24 h of treatment (Table S4). This increase correlates well with the induction of an early apoptosis in the case of (3) or secondary apoptosis in the cases of (3) and (4) compounds, respectively.
Conclusions
The cobalt(II), nickel(II), copper(II) and zinc(II) complexes with Schiff base 2-[(E)-1H-1,2,4-triazol-3-ylimino)methyl]phenol have been synthesised with good yields by template condensation.
The modifications in the IR spectra of complexes are in accordance with the condensation process and Schiff base coordination through azomethinic nitrogen and phenolic oxygen, respectively. Electronic spectrum of Ni(II) complex is characteristic for the hexacoordinate species, while that of Co(II) and Cu(II) complexes display the pattern of tetracoordinate ones. These data were furthermore confirmed for Co(II) and Ni(II) by magnetic behaviour of complexes at room temperature. The powder EPR spectrum as well as magnetic moment at room temperature sustains a dimeric structure for Cu(II) complex.
Cyclic voltammetric studies of the complexes emphasised their redox behaviour. The shifting of characteristic reduction peaks to more negative potentials for complexes in comparison with that of solvated species indicates that the Schiff base increases the complexes stability.
Thermal decomposition of complexes provides useful informations concerning the number and nature of water molecules, the composition of complexes and also the intervals of thermal stability. The thermal degradation occurs in three steps and comprises water and chloride elimination as well as Schiff base oxidative degradation. The Cu(II) complex has a different behaviour and melt after chloride anion elimination. The water elimination up to 145 °C accounts for its crystallisation nature in the case of Co(II) and Zn(II) complexes, while for Ni(II) complex, the coordinative nature of this species is related to the higher temperature of its release up to 170 °C. The results are in good concordance with the complexes composition.
The temperature corresponding to the decomposition of the anhydrous complexes increases in order of (1) = (4) < (2) < (3) indicating that Co(II) and Zn(II) compounds are the most covalent from this series.
The complexes exhibited an improved antimicrobial activity against both planktonic and biofilm-embedded cells in comparison with the ligand. This behaviour could be related to the known stereochemical versatility of these transition ions that allowed a good interaction with the target biomolecules.
The most active compounds against planktonic cells were the Cu(II) and Zn(II) complexes, while Co(II) complex inhibited the biofilm formed by the majority of the tested strains, demonstrating its utility for the design of novel materials and strategies for fighting against medical biofilms and associated chronic infections.
The Cu(II) and Zn(II) compounds proved to be also cytotoxic on the tumour cell lines, indicating another potential application for the biomedical filed, represented by the development of new anti-tumor agents.
The good biological activity of Cu(II) and Zn(II) complexes can be related to their stereochemical versatility. Furthermore, for Cu(II) species, the ability to easily change the oxidation state indicated by cyclic voltammetry and for Zn(II) complex the higher covalent degree indicated by thermal decomposition pattern can be also taken into account.
References
Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–22.
Lazar V. Quorum sensing in biofilms—how to destroy the bacterial citadels or their cohesion/power? Anaerobe. 2011;17:280–5.
Glinel K, Thebault P, Humblot V, Pradier CM, Jouenne T. Antibacterial surfaces developed from bio-inspired approaches. Acta Biomater. 2012;8:1670–84.
Tournu H, Van Dijck P. Candida Biofilms and the host: models and new concepts for eradication. Intern J Microbiol. 2012;2012:ID. 845352 doi:10.1155/2012/845352.
Potts KT. The Chemistry of 1,2,4-Triazoles. Chem Rev. 1961;61:87–127.
Agalave SG, Maujan SR, Pore VS. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem Asian J. 2011;6:2696–718.
Whiting M, Muldoon J, Lin Y-C, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew Chem Int Ed. 2006;45:1435–9.
Zhou C-H, Wang Y. Recent researches in triazole compounds as medicinal drugs. Curr Med Chem. 2012;19:239–80.
Sun N, Xie Y, Sheng C, Cao Y, Zhang W, Chen H, Fan G. In vivo pharmacokinetics and in vitro antifungal activity of iodiconazole, a new triazole, determined by microdialysis sampling. Int J Antimicrob Agents. 2013;41:229–35.
Kleemann A, Engel J. Pharmaceutical Substances. Stuttgart: Thieme Verlag; 1999.
Kharb R, Sharma PC, Yar MS. Pharmacological significance of triazole scaffold. J Enz Inhib Med Chem. 2011;26:1–21.
Sahoo S, Veliyath SK, Mahendra Kumar CB. Review on substituted 1,2,4-triazoles as potent antifungal and antibacterial agents. Int J Res Pharm Sci. 2012;3:326–33.
Wahi AK, Singh AK, Singh A. Design and synthesis of novel Schiff’s bases having N-(4H-1,2,4-triazole-4-yl)benzamido moiety as antimicrobial and anti-inflammatory agents. Pharm Chem. 2011;3:146–54.
Chandramouli C, Shivanand MR, Nayanbhai TB, Bheemachari B, Udupi RH. Synthesis and biological screening of certain new triazole schiff bases and their derivatives bearing substituted benzothiazole moiety. J Chem Pharm Res. 2012;4:1151–9.
Sumangala V, Poojary B, Chidananda N, Arulmoli T, Shenoy S. Synthesis and biological evaluation of some Schiff bases of 4-amino-5-(4-methylsulfonyl)benzyl-2,4-dihydro-3H-[1, 2, 4]-triazole-3-thione. Med Chem Res. 2013;22:2921–8.
Murthy YLN, Govindh B, Diwakar BS, Nagalakshmi K, Rao KVR. Synthesis and bioevaluation of Schiff and Mannich bases of isatin derivatives with 4-amino-5-benzyl-2,4-dihydro-3H-1,2,4-triazole-3-thione. Med Chem Res. 2012;21:3104–10.
Zheng Y, Xue W, Guo Q, Lu P, Wang Z, Yuan K. Synthesis and antitumor activity of 5,6-2H-[1,2,4]-triazolo[3,4-b][1,3,4]thiadiazine derivatives. Chin J Org Chem. 2011;31:912–6.
Serbest K, Özen A, Ünver Y, Er M, Değirmencioğlu I, Sancak K. Spectroscopic and theoretical study of 1,2,4-triazole-3-one based salicylaldimine complexes and evaluation of superoxide-scavenging properties. J Mol Struct. 2009;922:1–10.
Ma L, Lu L, Zhu M, Wang Q, Li Y, Xing S, Fu X, Gao Z, Dong Y. Mononuclear copper(II) complexes with 3,5-substituted-4-salicylidene-amino-3,5-dimethyl-1,2,4-triazole: Synthesis, structure and potent inhibition of protein tyrosine phosphatases. Dalton Trans. 2011;40:6532–40.
Kulkarni NV, Budagumpi S, Kurdekar GS, Revankar VR, Didagi S. Anticonvulsant activity and toxicity evaluation of Cu(II) and Zn(II) metal complexes derived from triazole-quinoline ligands. Chem Pharm Bull. 2010;58:1569–75.
Bagihalli GB, Avaji PG, Patil SA, Badami PS. Synthesis, spectral characterization, in vitro antibacterial, antifungal and cytotoxic activities of Co(II), Ni(II) and Cu(II) complexes with 1,2,4-triazole Schiff bases. Eur J Med Chem. 2008;43:2639–49.
Reddy V, Patil N, Reddy T, Angadi SD. Synthesis, characterization and biological activities of Cu(II), Co(II), Ni(II), Mn(II) and Fe(III) complexes with Schiff base derived from 3-(4-chlorophenoxymethyl)-4-amino-5-mercapto-1,2,4-triazole. Eur J Chem. 2008;5:529–38.
Zabin SA. Synthesis and antimicrobial studies of Cu(II), Ni(II) and Zn(II) Schiff base complexes derived from substituted 1,2,4-triazoles and heteroaromatic aldehydes. Asian J Chem. 2011;23:4067–71.
Chohan ZH, Hanif M. Synthesis and characterization of biologically active new Schiff bases containing 3-functionalized 1,2,4-triazoles and their zinc(II) complexes: crystal structure of 4-bromo-2-[(E)-(1H-1,2,4-triazol-3-ylimino)-methyl]phenol. Appl Organomet Chem. 2011;25:753–60.
Creaven BS, Devereux M, Foltyn A, McClean S, Rosair G, Thangella VR, Walsh M. Quinolin-2(1H)-one-triazole derived Schiff bases and their Cu(II) and Zn(II) complexes: possible new therapeutic agents. Polyhedron. 2010;29:813–22.
Altalbawy FMA, Mohamed GG, Sayed MAEE, Mohamed MIA. Synthesis, characterization, and biological activity of some transition metal complexes with Schiff base ligands derived from 4-amino-5-phenyl-4H-1,2,4-triazole-3-thiol and salicaldehyde. Monatsh Chem. 2012;143:79–89.
Hanif M, Chohan ZH. Synthesis, spectral characterization and biological studies of transition metal(II) complexes with triazole Schiff bases. Appl Organomet Chem. 2012;27:36–44.
Hanif M, Chohan ZH. Design, spectral characterization and biological studies of transition metal(II) complexes with triazole Schiff bases. Spectrochim Acta A Mol Biomol Spectrosc. 2013;104:468–76.
McCutcheon AR, Ellis SM, Hancock RE, Towers GH. Antibiotic screening of medicinal plants of the British Columbian native peoples. J Ethnopharmacol. 1992;37:213–23.
Marutescu L, Limban C, Chifiriuc MC, Missir A-V, Chirita IC, Caproiu MT. Studies on the antimicrobial activity of new compounds containing thiourea function. Biointerface Res Appl Chem. 2011;1:236–41.
Olar R, Badea M, Marinescu D, Chifiriuc MC, Bleotu C, Grecu MN, Iorgulescu EE, Lazar V. N, N-dimethylbiguanide complexes displaying low cytotoxicity as potential large spectrum antimicrobial agents. Eur J Med Chem. 2010;45:3027–34.
Awtar K. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–93.
Hawley RG, Hawley TS. Flow cytometry protocols. 2nd ed. New York: Humana Press; 2004.
Hassan FSM, Mohamed AD, Gabr AA, Gad AA. The preparation and characterization of divalent copper, nickel, cobalt and manganese complexes of some Schiff base ligands. AU. Sci. 1994;6:125–34.
Singh K, Kumar Y, Puri P, Kumar M, Sharma C. Cobalt, nickel, copper and zinc complexes with 1,3-diphenyl-1H-pyrazole-4-carboxaldehyde Schiff bases: antimicrobial, spectroscopic, thermal and fluorescence studies. Eur J Med Chem. 2012;52:313–21.
Abdel-Latif SA, Hassib HB, Issa YM. Studies on some salicylaldehyde Schiff base derivatives and their complexes with Cr(III), Mn(II), Fe(III), Ni(II) and Cu(II). Spectrochim Acta A. 2007;67:950–7.
Rathore K, Singh RKR, Singh HB. Structural, spectroscopic and biological aspects of O, N- donor Schiff base ligand and its Cr(III), Co(II), Ni(II) and Cu(II) complexes synthesized through green chemical approach. E-J Chem. 2010;7:566–72.
Nakamoto K. Infrared and Raman spectra of inorganic and coordination compounds. New York: Wiley; 1986.
Chohan ZH, Hanif M. Syhthesis and characterization of biologically active new Schiff bases containing 3-functionalized 1,2,4-triazoles and their zinc(II) complexes: crystal structure of 4-bromo-2-[(E)-(1H-1,2,4-triazol-3-ylimino)-methyl]phenol. Appl Organomet Chem. 2011;25:753–60.
Lever ABP. Inorganic electronic spectroscopy, Amsterdam, London. New York: Elsevier; 1986.
Gispert JR. Coordination chemistry. Weinheim: Wiley; 2008.
Kokoszka GF, Duerst RW. EPR studies of exchange coupled metal ions. Coord Chem Rev. 1970;5:209–44.
Zanello P. Inorganic electrochemistry: theory, practice and application. Cambridge: Royal Society of Chemistry; 2003.
Tatucu M, Rotaru P, Rau I, Spinu C, Kriza A. Thermal behaviour and spectroscopic investigation of some methyl 2-pyridyl ketone complexes. J Therm Anal Calorim. 2010;100:1107–14.
Vecchio S, Materazzi S, Wo LW, De Angelis Curtis S. Thermoanalytical study of imidazole-substituted coordinationcompounds: cu(II)-and Zn(II)-complexes of bis(1-methylimidazol-2-yl)ketone. Thermochim Acta. 2013;568:31–7.
Zianna A, Vecchio S, Gdaniec M, Czapik A, Hatzidimitriou A, Lalia-Kantouri M. Synthesis, thermal analysis, and spectroscopic and structural characterizations of zinc(II) complexes with salicylaldehydes. J Therm Anal Calorim. 2013;112:455–64.
Bujdošová Z, Gyoryová K, Mudroňová D, Hudecová D, Kovářová J. Thermoanalytical investigation and biological properties of zinc(II) 4-chloro-and 5-chlorosalicylates with N-donor ligands. J Therm Anal Calorim. 2012;110:167–76.
Findoráková L, Gyoryová K, Hudecová D, Mudronňvá D, Kovárŏvá J, Homzová K, El-Dien FAN. Thermal decomposition study and biological characterization of zinc(II) 2-chlorobenzoate complexes with bioactive ligands. J Therm Anal Calorim. 2013;111:1771–81.
Carvalho CT, Caires FJ, Lima LS, Ionashiro M. Thermal investigation of solid 2-methoxycinnamylidenepyruvate of some bivalent transition metal ions. J Therm Anal Calorim. 2012;107:863–8.
Yousef TA. Abu El-Reash, El-Gammal OA, Bedier RA. Co(II), Cu(II), Cd(II), Fe(II) and U(VI) complexes containing a NSNO donor ligand: synthesis, characterization, optical band gap, in vitro antimicrobial and DNA cleavage studies. J Mol Struct. 2012;1029:149–60.
Holló B, Krstić M, Sovilj SP, Pokol G, Mészáros Szécsényi K. Thermal decomposition of new ruthenium(II) complexes containing N-alkylphenothiazines. J Therm Anal Calorim. 2011;105:27–32.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Calu, L., Badea, M., Chifiriuc, M.C. et al. Synthesis, spectral, thermal, magnetic and biological characterization of Co(II), Ni(II), Cu(II) and Zn(II) complexes with a Schiff base bearing a 1,2,4-triazole pharmacophore. J Therm Anal Calorim 120, 375–386 (2015). https://doi.org/10.1007/s10973-014-3970-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-014-3970-5