
Abstract
Poly(l-lactide) (PLLA) and functionalized multi-walled carbon nanotubes (f-MWNTs) were used to prepare PLLA/f-MWNTs composites via solution blending. The structure and morphology of f-MWNTs were characterized using FT-IR and SEM. The spherulitic morphologies, isothermal crystallization kinetics, and melting behavior of the resulting PLLA/f-MWNTs composites were investigated by POM and DSC, respectively. Both Avrami and Lauritzen–Hoffman kinetics models are used to quantitatively evaluate the crystallization half-time t 1/2, the nucleation constant K g, and the work of chain folding q of PLLA and its composites. Temperature modulated DSC was used to investigate the mechanism of overlapped endothermic and exothermic peaks of PLLA/f-MWNTs composites. The results indicated that the SiO2 coating on the MWNTs could react with coupling agent KH-550 leading to the formation of f-MWNTs, which can be evenly dispersed in PLLA matrix. A decrease of spherulite size and an increase of crystallization rate were observed from POM measurements for PLLA/f-MWNTs. The multiple melting behavior can be attributed to the melt-recrystallization process of PLLA/f-MWNTs composites at certain temperature.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biodegradable polymers had attracted great interest in scientific community considering the environmental pollution from petroleum-based plastics [1]. Among various biodegradable plastics, poly(lactide) (PLA) is undoubtedly one of the most promising candidates with potential application on packaging and tissue engineering due to its biocompatibility and biodegradation [2–4]. PLA is produced either by ring-opening polymerization of lactide or by polycondension of lactic acid monomers, which can be obtained from fermentation of corn [5] and has been commercially available [6]. However, PLA has some limitations, such as slow crystallization rate, low crystalline degree, low-heating distortion temperature, and comparatively brittle and stiff [7], which greatly limit its practical applications [8, 9]. In order to overcome those drawbackes, several approaches have been proposed to improve PLA crystallization capabilities, including thermomechanical treatments, plasticizers, as well as, nucleating agents. Another route to enhance PLA mechanical properties is to mix with more or less miscible polymers or with nanofillers [10–15]. For example, nanoparticles such as organically modified layered nano silicate clay particles (clay), zinc phenylphosphonate (PPZn), silica nanoparticles (SiO2), oxidized multi-walled carbon nanotubes (O-MWCNTs), organo-montmorillonite (OMMT), and so on had been used to enhance the crystallization rate and mechanical properties of PLA [16–22].
Carbon nanotubes (CNTs), as an ideal modification material, have attracted considerable attention due to their unique and remarkable physical and mechanical properties [23–25]. It was reported that the strength, thermostability, crystallinity, and electrical conductivity of polymer/CNTs composites can be enhanced simultaneously due to the unique characteristics of CNTs [26–29]. Those properties observed at nanoscale have motivated many researchers to utilize CNTs as a new reinforcement in polymer composites [30]. PLA/CNTs nanocomposites, the notable example of biocomposites, attracted increasing attention because of their biodegradability and biocompatibility [31, 32], which may be of great significant for its wide practical application.
However, high L/D ratio and small size affect make CNTs agglomerate very easily. Thus, surface functionalization of CNTs is desired, which can be achieved via both covalent and non-covalent reactions with encapsulating polymer layer, organic or amphiphilic molecules, ionic moieties. The functionalization process is believed to reduce the interfacial tension between the polymer matrix and CNTs to enhance dispersion and interfacial adhesion [33].
As known, both mechanical properties and degradation rate of PLA were signficantly influenced by its crystalline morphology. It is very important to investigate the crystalline behavior of PLA-based composites. Xu et al. studied the non-isothermal crystallization of PLA/MWNTs composite and found that MWNTs could act as a nucleating agent to enhance nucleation of crystallization [34]. Wu et al. investigated nucleation mechanisms and the crystallization kinetics of PLA/CNT composites. The results showed that nanotubes have nucleating effect on both the melt crystallization and the cold crystallization of PLA [35]. Barras et al. [36] used X-ray diffraction to prove that PLA crystal structure is not modified by the presence of CNTs.
Having such excellent comprehensive properties simultaneously as toughening, reinforcing, and accelerating crystallization effects et al., MWNTs was selected as a nucleating agent for PLLA in this article to investigate the influence of functionalized MWNTs on spherulitic morphologies, isothermal crystallization kinetics, and melting behavior of PLLA/f-MWNTs by using temperature modulated DSC. The relationship between structure and properties of biodegradable PLLA/f-MWNTs composites was discussed.
Experimental
Materials
PLLA pellets (REVODE101, M n = 50,000–60,000) and MWNTs (purity >95 %, outer diameter 10–20 nm, length 5–15 μm) were kindly provided by Hisun Biomaterials Co. Ltd. (Zhejiang, China) and NanotechPort Co. Ltd. (Shenzhen, China), respectively. 3-aminopropyltriethoxysilane (KH-550) and sodium dodecyl sulfonate (SDS) were purchased from Lanteng Chemical Technology INC. and Yongda Chemical Reagent Inc. (Tianjin, China), respectively. Tetraethoxysilane (TEOS), concentrated nitric acid and sulfuric acid were obtained from North Tianyi Chemical Reagent Inc. (Tianjin, China).
Modification of MWNTs
Modification of MWNTs was carried out according to the literatures [18, 37]. Equal amount of purified MWNTs and SDS was mixed in 100 fold water at room temperature and ultrasonicated for 1 h to obtain a homogeneous dispersion. 10 mL mixed solvent containing TEOS, deionized water, and anhydrous ethanol with a volume ratio of 2:1:5 was added slowly into 400 mL deionized water with well-dispersed MWNTs under magnetic stirring. After 48 h at room temperature, SiO2 coated MWNTs (SiO2-MWNTs) were obtained. The coupling agent KH-550 of 1 and 1 g SiO2-MWNTs was mixed in anhydrous ethanol (400 mL) under magnetic stirring at 70 °C for 24 h. The functionalized MWNTs (f-MWNTs) could be obtained through the reaction between KH-550 and SiO2-MWNTs.
Preparation of PLLA/f-MWNTs composites
PLLA of 1 g was dissolved in 30 mL of chloroform and mixed with different amount of f-MWNTs. A thin film of each sample was obtained by solution casting. Prior to the characterization, the samples were dried at 80 °C for 12 h in a vacuum oven.
Characterization
Fourier transform infrared spectroscopy (FTIR)
Infrared spectra of the functionalized MWNTs were collected using a Bruker VECTOR-22 spectrometer. The MWNTs were dried at 80 °C for 12 h, and then KBr pellets were prepared with the dried MWNTs. The infrared spectrum was recorded in the scanning range of 400 to 4,000 cm−1.
Scanning electron microscopy (SEM)
The morphologies of MWNTs and the fracture surface of PLLA/f-MWNTs composite specimens were analyzed with a JEOL JSM-6380LV scanning electron microscope (SEM) with an acceleration voltage of 30 kV. The fractured surfaces of specimens were sputter coated with a thin layer of gold and then examined for morphological structure.
Differential scanning calorimetry (DSC)
Dynamic DSC experiments were conducted in a NETZSCH 204F1 differential scanning calorimeter operating in a nitrogen environment. Samples of 5–10 mg sealed in aluminum pans were first heated to 200 °C for 2 min to eliminate the thermal history, and then cooled at a rate of 80 °C min−1 to predetermined crystallization temperature (T c) for 30 min. The samples were subsequently reheated to 200 °C at a rate of 10 °C min−1. During DSC scanning, the temporal development of crystallinity and melting temperature (T m) at various T c was recorded.
Temperature modulated mode (TM-DSC) was used to investigated the multiple crystallization and melting peaks of PLLA/f-MWNTs (0.1 mass%) composite. During the test, the specimen was first heated to 200 °C for 2 min to eliminate thermal history, and then cooled at a rate of 80 °C min−1 to 100 °C for 30 min to crystallize. The specimen was reheated to 200 °C at a rate of 5 °C min−1 with the modulation for temperature. The special parameter, period and amplitude, were 40 s and 0.5 k, respectively.
Polarized optical microscopy (POM)
The morphology of spherulites was monitored with a polarized optical microscopy (Caikon XP-500 China). A thin film of each sample obtained by solution casting was inserted between two microscope cover slides and heated to 200 °C to molten state, then quickly transferred to a hot stage and equilibrated at the desired isothermal crystallization temperature (T c) to isothermally crystallize, and monitored the spherulite growth and taken photographs.
Results and discussion
Characterization of f-MWNTs
Figure 1 shows the infrared spectra of p-MWNTs, SiO2-MWNTs, and f-MWNTs. The absorption peaks at 1,637 and 3,450 cm−1 are attributed to Si–OH bending vibration and symmetrical stretching vibration, respectively. The peaks at 464, 798, and 1,099 cm−1 are the absorption for Si–O–Si bending vibration, symmetrical and antisymmetric stretching vibration, respectively. These five characteristic peaks of SiO2-MWNTs indicate that the MWNTs have been coated by a layer of SiO2, because of the hydrolysis of TEOS.

FT-IR spectra of p-MWNTs, SiO2-MWNTs, and f-MWNTs
Furthermore, there is an apparent absorption peak at 2,930 cm−1 for the spectra of f-MWNTs, which is attributed to the stretching vibration of –CH2– in KH-550 and its hydrolysate. This demonstrates that KH-550 has reacted with the SiO2-MWNTs, and SiO2-MWNTs were grafted coupling agent KH-550.
The KH-550 coupling agent hydrolyzed first and got the silanol, which can react with the hydroxy on the surface of the SiO2-MWNTs forming a thin film on the surface of the SiO2-MWNTs. Alkyl group of the coupling agent has compatibility with the PLLA matrix and so the f-MWNTs can combine with PLLA tightly.
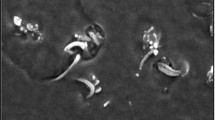
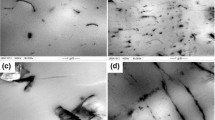
Figure 2 gives SEM images of p-MWNTs, f-MWNTs, and fracture surface of PLLA/f-MWNTs composite. As can be seen from Fig. 2a, the p-MWNTs aggregated severely. While f-MWNTs can homogeneously disperse owing to surface modification (cf. Fig. 2b), and the resulting f-MWNTs wouldn’t aggregate again into PLLA (cf. Fig. 2c). This homogeneous dispersion should be attributed to the improvement of interfacial action between the f-MWNTs and PLLA matrix. This demonstrates that strong interaction exists between f-MWNTs and PLLA chains, which facilitates the high dispersing ability of f-MWNTs in PLLA media [38].

SEM images of a p-MWNTs, b f-MWNTs and c the fracture surface of PLLA/0.5 mass% MWNTs composite
Isothermal crystallization kinetics
The isothermal crystallization curves of PLLA and PLLA/0.5 mass% f-MWNTs composite at various crystallization temperatures (T c) are shown in Fig. 3a and b. A minimum of crystallization exothermic peak position was obtained at 105 °C. As T c increases, the crystallization exothermic peak shifts to a lower value before 105 °C and to a higher value after 105 °C.

a, b are heat flow versus t for neat PLLA & PLLA/0.5 mass% f-MWNTs, respectively, during isothermal crystallization at the designated temperature; c is isothermal crystallization curve of all sample during T c = 105 °C
Figure 3c shows isothermal crystallization curve of all samples at T c = 105 °C. Compared to PLLA/f-MWNTs composites, there is no apparent exothermic peak for neat PLLA, which means neat PLLA is very difficult to crystallize without nucleating agent. The crystallization of neat PLLA is strongly affected by f-MWNTs addition.
The addition of f-MWNTs shortens the time to reach the crystallization halftime, indicating a faster crystallization rate, as shown in Table 1. This result suggests that during the isothermal crystallization, f-MWNTs acts as a nucleating agent and contributes to an improvement in the crystallinity of PLLA composites.
Based on the curves in Fig. 3, the isothermal crystallization kinetics can be analyzed by evaluating the relative crystallinity (X t ) versus crystallization time (t) at the designated temperature, which can be calculated according to the following equation:
where dH c /dt is the rate of heat evolution. X t can be obtained from the area of the exothermic peak for isothermal crystallization as shown in Fig. 3. Figure 4a and b shows the plots of X t versus t of neat PLLA and PLLA/0.5 mass% f-MWNTs composite at different crystallization temperatures. The characteristic sigmoidal isothermals roughly shift to the left along the time axis with the crystallization temperature comes close to 105 °C, which indicates that crystallization rate increases. As shown in Fig. 4c, the fact that X t of the PLLA/f-MWNTs composites is higher than that of neat PLLA, suggesting that the f-MWNTs can increase the rate of crystallization for PLLA.

a, b are plots of X t versus t of neat PLLA and PLLA/0.5 mass% f-MWNTs composite, respectively, during isothermal crystallization at certain temperature; c is X t versus t for isothermal crystallization of all samples at T c = 105 °C
Avrami kinetics theory model
Avrami equation can be employed to analyze the isothermal crystallization kinetics of PLLA and PLLA/f-MWNTs composites. It assumes that the relative degree of crystallinity X t develops as a function of crystallization time t as follows:
For practical purpose, Eq. (2) is usually rearranged in its double logarithmic form as Eq. (3):
where X t is the relative crystallinity at time t, k and n values devote the crystallization rate constant and the Avrami exponent, respectively. Plotting ln[−ln(1 − X t)] versus lnt, as shown in Fig. 5, the values of k and n can be obtained from the intercept and slope of the starting linear portion, respectively. The crystallization half-time (t 1/2), defined as the time when X t is 50 %, is the most important parameter of isothermal crystallization kinetics, and it can be calculated as follows:
Generally, the growth rate (G) is defined as the reciprocal of t 1/2 (G = 1/t 1/2), which is used to characterize the crystallization rate. And the four parameters (k, n, t 1/2, G) of PLLA and PLLA/f-MWNTs composites versus different T c are listed in Table 1.

a, b are plots of ln[−ln(1 − X t)] versus t of neat PLLA and PLLA/0.5 mass% f-MWNTs composite, respectively, during isothermal crystallization at certain temperature; c is ln[−ln(1 − X t)] versus t for isothermal crystallization of all samples at T c = 105 °C
The Avrami exponent n reveals the nucleation mechanism and growth dimension of polymers [39]. The values of n for PLLA and PLLA/f-MWNTs composites mostly ranged from 2.0 to 2.6, indicating that the crystal morphology corresponds to a mixed two- and three-dimension growth. With the ratio of f-MWNTs increasing, the values of n decrease obviously and come close to 2.0. The space of crystal growth is blocked by f-MWNTs, leading to the decrease of n. The f-MWNTs in PLLA may cause heterogeneous nucleation and lead to the transformation in the crystal growth from two- and three-dimension growth to two-dimension growth.
The crystallization rate constant k is a parameter to characterize the nucleation rate and growth process. The values of k increase obviously due to f-MWNTs, so that the crystallization rate can be enhanced. However, the excess f-MWNTs may hinder the movement of chain segment, leading to a decrease of crystallization rate.
Lauritzen–Hoffman kinetics model
The Lauritzen–Hoffman equation is often used to analyze the temperature dependence of the spherulites growth rate G of neat PLLA and is composites [40].
For convenience, Eq. (5) can be rewritten as follows:
where G 0 denote the growth rate of crystallization and pre-exponential factor that includes all temperature-dependent terms; R denotes the universal gas constant; U* is the diffusional for the transport of segments to the crystallizable site at the liquid- solid interface taken as 6,280 J/mol; ΔT is the degree of supercooling, defined as T 0m − T c (T 0m can be evaluated via analyzing the calorimetric results [41]); T ∞ is the hypothetical temperature where all motion associated with viscos flow ceases, which is usually assumed to be equal to (T g − 30)K, and the term f = 2 T c/(T 0m + T c) is the correction factor that accounts for the changes in heat of fusion as the temperature; T g and K g are glass transition temperature and the nucleation parameter related to surface free energy.
Plotting lgG + U*/[R(T c − T ∞ )] versus 10−4/(T cΔT f), shown as in Fig. 6, lnG 0 and −K g can be obtained from the intercept and slope of the straight line, respectively. The values of K g are shown in Table 2. The nucleation constant K g for neat PLLA is lower than those for PLLA/f-MWNTs composites.

Plots of ln G + U*/R(T c − T ∞ ) versus 105/T cΔT f for isothermal crystallization of PLLA and PLLA/f-MWNTs composites
The nucleation constant K g is related to the surface free energy, and the relation can be pressed by the following equation:
where σ and σ e are the lateral (side surface) and fold surface free energies of the growing crystal, respectively, and the value of σ is 1.54 × 10−2J m−2; b 0 is the layer thickness of the single molecular layer (stem) in the crystal, which is 5.14 × 10−10m; k B is the Boltzmann constant; ΔH 0m is the heat fusion per unit volume, with a recommended value of ΔH 0m = 1.11 × 108 J m−2. n is a variable parameter depending on the crystallization regime, and theoretically assumed the value of 4 for the regimes I and III, and the value of 2 for the regime II, respectively.
The Lauritzen approximated Z-test equation defined as follows can be used to determine the regime in which the crystallization occurs (in either I or II):
where L is the crystalline substrate length. In Eq. (8), X = K g, Z < 0.1 if the crystallization growth occurs in regime I, but for regime II, X = 2 K g, Z > 1. Whether the calculation for L is reasonable can be used to judge which of regime I and II is available [42]. Testing for regime I, the values of L should be <0.56–1.64 nm for the five sample, and those are all unrealistically small. However, for regime II, L should be >97–822 nm, which is the appropriate range for L. Thus, the crystallization growth of PLLA and PLLA/f-MWNTs composites proceeded according to regime II.
For regime II, the values of the fold surface free energy (σ e) are calculated according to Eq. (6). σ is often estimated by Thomas–Stavely empirical relation as follows [43]:
where a 0 and b 0 are the width and thickness of the stem added on the substrate whose values are 5.97 × 10−10 and 5.17 × 10−10 m, respectively. The value of α for organic molecules is closer to 0.30, and α = 0.25 has been chosen for estimation the surface free energy (α) in the present case as appropriate to high melting polyesters [44]. For PLLA, σ is estimated as 1.54 × 10−2 J m−2 using ΔH 0m = 1.11 × 108 J m−2.
The values of K g and σ e obtained from Eq. (6) are illustrated in Table 2. Obviously, the values of σ e for PLLA/f-MWNTs composites are higher than that for neat PLLA. The σ e present the work of chain folding, which suggests that there exist stronger constraints on the mobility of the polymer chains in the interspherulitic regions for PLLA/f-MWNTs composites than that for neat PLLA [45].
Furthermore, the work of chain folding q summarized in Table 2 can be calculated approximately from Eq. (10):
The higher q value in composites could be ascribed to a general constraint of the PLLA chain mobility in the composites melt caused by f-MWNTs [45], which improved the ability of nucleation.
POM investigation
Figure 7 shows the POM micrographs of PLLA and PLLA/f-MWNTs composites, and the spherulites morphology can be observed directly. All the micrographs present the typical Maltese-cross spherulites. The spherulites size decreases distinctly with the content of f-MWNTs increasing. Based on these observed POM images, the nucleation density and nucleation rates of PLLA/1 mass% MWNTs composites are higher than that of PLLA, which is consistent with the DSC data. These results revealed that good dispersion of MWNTs can effectively accelerate the crystallization development and change the crystallization kinetics of PLLA. As a nucleating agent, good dispersibility of MWNTs in PLLA matrix was benefit to nuclei formation and enhanced crystallization rate.

POM images of PLLA and PLLA/f-MWNTs isothermally crystallized at 105 °C for 2 h: a neat PLLA; b PLLA/0.1 mass% f-MWNTs; c PLLA/0.5 mass% f-MWNTs; d PLLA/1.0 mass% f-MWNTs
Melting behavior
The second heating curves of PLLA and PLLA/f-MWNTs composites annealed at the constant T c are shown in Fig. 8. The melting curves of PLLA and PLLA/f-MWNTs composite have the same shape and variation trend, indicating that the addition of f-MWNTs cannot change the nucleation and growth mechanisms of crystallization for PLLA.

DSC curves of melting process for all samples crystallized at the specified crystallization temperature: a 95 °C; b 100 °C; c 105 °C; d 110 °C; e 115 °C; f 120 °C
The typical double melting peaks can be observed distinctly at the specified crystallization temperature between 95 and 115 °C as References [46, 47] and disappeared at 120 °C [48]. It is very obviously that peak I shifted to higher temperature and its magnitudes increases with increasing T c at 95 ≤ T c ≤ 115 °C (Fig. 8a–e). Meanwhile, the location of peak II remains almost unaltered. These distinct melting behaviors are analogous with some earlier reports for melt-crystallized PLLA with a slight difference in temperature range [49, 50].
The multiple-melting behavior is caused by the melt-recrystallization process [46] and polymorphism and crystallization [48]. The initial imperfect crystals formed during specified isothermal crystallization are molten at a lower temperature and reorganized to the more stable and perfect crystals molten at a higher temperature. The double peaks correspond to the melting behavior of initial imperfect crystals and reorganized perfect crystals, respectively. The higher the crystallization temperature, the higher of the relative crystallinity, the more stable of the initial crystals, the higher melting temperature corresponding the peak I. PLLA and its composites can form so perfect and stable crystals at 120 °C that there is no melt-recrystallization process and the double melting peaks disappeared.
The observation of multiple-melting endotherms is a common phenomenon for many semicrystalline polymers, being observed for several other polyesters, including poly(ethylene terephthalate) (PET), poly-(butylene terephthalate) (PBT), poly(3-hydroxybutyrate) (PHB), and its copolymers with 3-hydroxyvalerate (PHBV) [48]. It is an important research subject in the field of polymer science to gain a basic understanding of the structural evolution related to thermal history (i.e., crystallization conditions) and to provide a deep insight into the crystallization and melting processes of semicrystalline polymers. So far no consensus has been reached as for the exact origin of multiple-melting behavior of PLLA [51–53]. This behavior can, in principle, be ascribed to a number of factors, such as the presence of more than one crystal modification, molecular weight segregation that accompanies crystallization, different crystal morphologies, or melting-recrystallization-remelting processes that occur during the DSC heating scan [54–56].
To better understand the mechanism of multiple melting behavior and melt-recrystallization process of PLLA and its composites, TM-DSC was used to separate the overlapped endothermic and exothermic peaks. Figure 9 is the TM-DSC curves of PLLA/0.1 mass% f-MWNTs composite at T c = 100 °C for 30 min. The solid line is superposition of the two dashed curves (reversing curve and non-reversing curve), which are the melting curves of initial crystals and recrystallization and melting curve, respectively.

TMDSC curves of melting for PLLA/0.1 mass% f-MWNTs composite isothermally crystallized at 100 °C
As reported [48], PLLA displays the multiple melting peaks in the DSC heating scan (Fig. 8) and crystalline polymorphism and crystallizes in α′, α, β, or γ form depending on the crystallization conditions. Each of these has distinctly different structural parameters. The α′ or α crystal is formed under most processing conditions depending on thermal history. Formed at lower temperatures than the α phase, most PLLA products are processed with experimental conditions favoring the α′ form. The α′ crystal has the similar chain conformation as α crystal but uniform conformational disorder in the Cα–C torsion angle, which leads to different crystalline forms with different thermal stabilities. Significantly different values for the two crystalline forms were obtained ΔH m(α′) = 57 ± 3 J g−1 and ΔH m(α) = 96 ± 3 J g−1 [57]. The melting temperature of α′ crystal is slightly lower than that of α crystal, and the less stable α′ crystal is very easy to transform into the more stable α phase during heating [48, 49, 57]. Previous studies found that dependent on the crystallization temperature (T c), the disorder (α′) and order (α) phases of PLLA are formed at low (T c < 100 °C) and high (T c ≥ 120 °C) temperatures, respectively [47].
From Fig. 9, it can be seen that there are three endothermic peaks (P 1, P 2 and P 3) and one exothermic peak (P exo). The melting temperature range of the PLLA and its composites is between 140 and 160 °C. Appearing in the same temperature range (about 140 °C) as P exo, P 1 is easy to be neglected in DSC scan. The corresponding temperature of P 2 and P 3 is about 160 °C. As mentioned above, α′ and α crystals are mainly formed at T c < 100 °C and T c ≥ 120 °C, respectively. It can be concluded that α′, α, and some imperfect crystals could be simultaneously formed at T c = 100 °C. The imperfect crystals formed upon primary crystallization have a greater tendency to reorganize into more stable structures (α phase) during the heating scan that leads to fusion. P 1 and P exo are attributed to the melting process and recrystallization of original imperfect crystals. P 2 and P 3 stand for the melting process of metastable crystals (α′ phase) and stable crystals (α phase), respectively.
The complex melting behavior of PLLA and its composites arises from the fusion of a certain amount of the original imperfect crystals (endothermic peak P 1), followed by recrystallization and final melting of more perfect crystals (exothermic peak P exo); the melting of metastable crystals α′ phase (endothermic peak P 2) and the fusion of stable crystals α already perfected during the heating scan (endothermic peak P 3).
Conclusions
Biodegradable PLLA/f-MWNTs nanocomposites with different f-MWNTs loading have been prepared successfully via solution casting method. A systematic research on the structure and morphology of f-MWNTs, spherulitic morphologies, isothermal crystallization kinetics, and melting behavior of the neat PLLA and its composites has been performed by using FTIR, SEM, POM, and DSC. FTIR results indicated that MWNTs coated with SiO2 could react with coupling agent KH-550. SEM images indicated a homogeneous dispersion of f-MWNTs throughout PLLA matrix. POM images show that f-MWNTs could act as nucleating agent leading to an decrease of spherulite size. The isothermal crystallization kinetics of neat PLLA improve the stability of nucleation for PLLA. The crystallization rate of PLLA is enhanced significantly due to the nucleation effect induced by f-MWNTs, while the crystallization mechanism does not change in the presence of f-MWNTs in the composites. The multiple melting behaviors can be attributed to the melt-recrystallization process of PLLA and PLLA/f-MWNTs composites at the designated temperature.
References
Dornburg V, Hermann BG, Patel MK. Scenario projections for future market potentials of biobased bulk chemicals. Environ Sci Technol. 2008;42(7):2261–7.
Reed A, Gilding D. Biodegradable polymers for use in surgery-poly (glycolic)/poly (lactic acid) homo and copolymers: 2: in vitro degradation. Polymer. 1981;22(4):494–8.
Kalb B, Pennings A. General crystallization behaviour of poly (l-lactic acid). Polymer. 1980;21(6):607–12.
Marega C, Marigo A, Di Noto V, Zannetti R, Martorana A, Paganetto G. Structure and crystallization kinetics of poly (l-lactic acid). Die Makromol Chem. 1992;193(7):1599–606.
Yu T, Ren J, Li S, Yuan H, Li Y. Effect of fiber surface-treatments on the properties of poly (lactic acid)/ramie composites. Compos A Appl Sci Manuf. 2010;41(4):499–505.
Kunioka M, Ninomiya F, Funabashi M. Biodegradation of poly (lactic acid) powders proposed as the reference test materials for the international standard of biodegradation evaluation methods. Polym Degrad Stab. 2006;91(9):1919–28.
Jacobsen S, Fritz H-G. Plasticizing polylactide—the effect of different plasticizers on the mechanical properties. Polym Eng Sci. 1999;39(7):1303–10.
Wang H, Qiu Z. Crystallization kinetics and morphology of biodegradable poly (l-lactic acid)/graphene oxide nanocomposites: influences of graphene oxide loading and crystallization temperature. Thermochim Acta. 2012;527:40–6.
Cohn D, Hotovely Salomon A. Designing biodegradable multiblock PCL/PLA thermoplastic elastomers. Biomaterials. 2005;26(15):2297–305.
Masirek R, Piorkowska E, Galeski A, Mucha M. Influence of thermal history on the nonisothermal crystallization of poly (l-lactide). J Appl Polym Sci. 2007;105(1):282–90.
Chrissafis K, Pavlidou E, Paraskevopoulos KM, Beslikas T, Nianias N, Bikiaris D. Enhancing mechanical and thermal properties of PLLA ligaments with fumed silica nanoparticles and montmorillonite. J Therm Anal Calorim. 2011;105:313–23.
Li H, Huneault MA. Effect of nucleation and plasticization on the crystallization of poly (lactic acid). Polymer. 2007;48(23):6855–66.
Chen H, Pyda M, Cebe P. Non-isothermal crystallization of PET/PLA blends. Thermochim Acta. 2009;492(1):61–6.
Fujimori A, Ninomiya N, Masuko T. Influence of dispersed organophilic montmorillonite at nanometer-scale on crystallization of poly (l-lactide). Polym Eng Sci. 2008;48(6):1103–11.
Bordes P, Pollet E, Avérous L. Nano-biocomposites: biodegradable polyester/nanoclay systems. Prog Polym Sci. 2009;34(2):125–55.
Day M, Nawaby AV, Liao X. A DSC study of the crystallization behavior of polylactic acid and its nanocomposites. J Therm Anal Calorim. 2006;86(3):623–9.
Pan P, Liang Z, Cao A, Inoue Y. Layered metal phosphonate reinforced poly (l-lactide) composites with a highly enhanced crystallization rate. ACS Appl Mater Interfaces. 2009;1(2):402–11.
Papageorgiou G, Achilias D, Nanaki S, Beslikas T, Bikiaris D. PLA nanocomposites: effect of filler type on non-isothermal crystallization. Thermochim Acta. 2010;511(1):129–39.
Zhao Y, Qiu Z, Yang W. Effect of functionalization of multiwalled nanotubes on the crystallization and hydrolytic degradation of biodegradable poly (l-lactide). J Phys Chem B. 2008;112(51):16461–8.
Pan H, Qiu Z. Biodegradable poly (l-lactide)/polyhedral oligomeric silsesquioxanes nanocomposites: enhanced crystallization, mechanical properties, and hydrolytic degradation. Macromolecules. 2010;43(3):1499–506.
Yu J, Qiu Z. Preparation and properties of biodegradable poly (l-lactide)/octamethyl-polyhedral oligomeric silsesquioxanes nanocomposites with enhanced crystallization rate via simple melt compounding. ACS Appl Mater Interfaces. 2011;3(3):890–7.
Chow WS, Lok SK. Thermal properties of poly(lactic acid)/organo-montmorillonite nanocomposites. J Therm Anal Calorim. 2009;95(2):627–32.
Salvetat J-P, Briggs GAD, Bonard J-M, Bacsa RR, Kulik AJ, Stöckli T, et al. Elastic and shear moduli of single-walled carbon nanotube ropes. Phys Rev Lett. 1999;82(5):944.
Qi YN, Xu F, Sun LX. Thermal stability and glass transition behavior of PANI/MWNT composites. J Therm Anal Calorim. 2008;94(1):137–41.
Subramoney S. Novel nanocarbons: structure, properties, and potential applications. Adv Mater. 1998;10(15):1157–71.
Rahmatpour A, Aalaie J. Steady shear rheological behavior, mechanical properties, and morphology of the polypropylene/carbon nanotube nanocomposites. J Macromol Sci B. 2008;47(5):929–41.
Kaganj AB, Rashidi AM, Arasteh R, Taghipoor S. Crystallisation behaviour and morphological characteristics of poly (propylene)/multi-walled carbon nanotube nanocomposites. J Exp Nanosci. 2009;4(1):21–34.
Sahoo NG, Cheng HKF, Cai J, Li L, Chan SH, Zhao J, et al. Improvement of mechanical and thermal properties of carbon nanotube composites through nanotube functionalization and processing methods. Mater Chem Phys. 2009;117(1):313–20.
Kim KH, Jo WH. A strategy for enhancement of mechanical and electrical properties of polycarbonate/multi-walled carbon nanotube composites. Carbon. 2009;47(4):1126–34.
Moniruzzaman M, Winey KI. Polymer nanocomposites containing carbon nanotubes. Macromolecules. 2006;39(16):5194–205.
Ray SS. Polylactide-based bionanocomposites: a promising class of hybrid materials. Acc Chem Res. 2012;45:1710–20.
Raquez JM, Habibi Y, Murariu M, Dubois P. Polylactide(PLA)-based nanocomposites. Prog Polym Sci. 2013;38:1504–42.
Wu D, Wu L, Zhang M, Zhao Y. Viscoelasticity and thermal stability of polylactide composites with various functionalized carbon nanotubes. Polym Degrad Stab. 2008;93(8):1577–84.
Xu Z, Niu Y, Yang L, Xie W, Li H, Gan Z, et al. Morphology, rheology and crystallization behavior of polylactide composites prepared through addition of five-armed star polylactide grafted multiwalled carbon nanotubes. Polymer. 2010;51(3):730–7.
Wu D, Wu L, Zhou W, Zhang M, Yang T. Crystallization and biodegradation of polylactide/carbon nanotube composites. Polym Eng Sci. 2010;50(9):1721–33.
Barrau S, Vanmansart C, Moreau M, Addad A, Stoclet G, Lefebvre J-M, et al. Crystallization behavior of carbon nanotube–polylactide nanocomposites. Macromolecules. 2011;44(16):6496–502.
Song W, Zheng Z, Tang W, Wang X. A facile approach to covalently functionalized carbon nanotubes with biocompatible polymer. Polymer. 2007;48(13):3658–63.
Neelgund GM, Oki A. Pd nanoparticles deposited on poly (lactic acid) grafted carbon nanotubes: synthesis, characterization and application in Heck C–C coupling reaction. Appl Catal A. 2011;399(1):154–60.
Huang J-W. Effect of nanoscale fully vulcanized acrylic rubber powders on crystallization of poly (butylene terephthalate): isothermal crystallization. Eur Polym J. 2007;43(10):4188–96.
Hoffman JD, Miller RL. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: theory and experiment. Polymer. 1997;38(13):3151–212.
Hoffman JD, Weeks JJ. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J Res Natl Bur Stand A. 1962;66(1):13–28.
Kalkar A, Deshpande V, Kulkarni M. Isothermal crystallization kinetics of poly (phenylene sulfide)/TLCP composites. Polym Eng Sci. 2009;49(2):397–417.
Di Lorenzo ML. Determination of spherulite growth rates of poly (l-lactic acid) using combined isothermal and non-isothermal procedures. Polymer. 2001;42(23):9441–6.
Hoffman JD. Theory of the substrate length in polymer crystallization: surface roughening as an inhibitor for substrate completion. Polymer. 1985;26(12):1763–78.
Cai J, Liu M, Wang L, Yao K, Li S, Xiong H. Isothermal crystallization kinetics of thermoplastic starch/poly (lactic acid) composites. Carbohydr Polym. 2011;86(2):941–7.
Pan P, Kai W, Zhu B, Dong T, Inoue Y. Polymorphous crystallization and multiple melting behavior of poly (l-lactide): molecular weight dependence. Macromolecules. 2007;40(19):6898–905.
Zhang J, Tashiro K, Tsuji H, Domb AJ. Disorder-to-order phase transition and multiple melting behavior of poly (l-lactide) investigated by simultaneous measurements of WAXD and DSC. Macromolecules. 2008;41(4):1352–7.
Di Lorenzo ML. Calorimetric analysis of the multiple melting behavior of poly(L-lactic acid). J Appl Polym Sci. 2006;100(4):3145–51.
Song P, Chen GY, Wei ZY. Calorimetric analysis of the multiple melting behavior of melt-crystallized poly(l-lactic acid) with a low optical purity. Therm Anal Calorim. 2013;111:1507–14.
Wang Y, Mano JF. Role of thermal history on the thermal behavior of poly(l-lactic acid) studied by DSC and optical microscopy. J Therm Anal Calorim. 2005;80:171–5.
Xu HS, Dai XJ, Lamb PR, Li ZM. Poly(l-lactide) crystallization induced by multiwall carbon nanotubes at very low loading. J Polym Sci B. 2009;47:2341–52.
Calafe M, Remiro PM, Cortázar MM, Calahorra ME. Cold crystallization and multiple melting behavior of poly(l-lactide) in homogeneous and in multiphasic epoxy blends. Colloid Polym Sci. 2010;288:283–96.
Shen C, Wang Y, Li M, Hu D. Crystal modifications and multiple melting behavior of poly(l-lactic acid-co-d-lactic acid). J Polym Sci B. 2011;49:409–13.
Liu T, Petermann J. Multiple melting behavior in isothermally cold crystallized isotactic polystyrene. Polymer. 2001;42:6453–61.
Gunaratne LMWK, Shanks RA. Multiple melting behaviour of poly(3-hydroxybutyrate-co-hydroxyvalerate) using step-scan DSC. Eur Polym J. 2005;41:2980–8.
Shan GF, Yang W, Tang XG. Multiple melting behaviour of annealed crystalline polymers. Polym Test. 2010;29:273–80.
Kalish JP, Aou K, Yang X. Spectroscopic and thermal analyses of α′ and α crystalline forms of poly(l-lactic acid). Polymer. 2011;52:814–21.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shi, J., Lu, X., Li, H. et al. Isothermal crystallization kinetics and melting behavior of PLLA/f-MWNTs composites. J Therm Anal Calorim 117, 1385–1396 (2014). https://doi.org/10.1007/s10973-014-3885-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-014-3885-1