
Abstract
LPSF/GQ-130 is a drug candidate, according to reports about its significant anti-inflammatory activity and non-toxicity demonstrated in an acute preclinical study. Despite this, knowledge of its physical–chemical properties is insufficient for the development of medicines. Thus, this work aimed to characterize the raw material at its molecular, particle, and agglomerate level as well as evaluate its thermal compatibility to pharmaceutical excipients. Through spectrometric techniques the molecular structure of the substance was confirmed. For thermal analysis its melting (171.3–176.5 °C) and degradation (238.3–297.4 °C) ranges, besides its purity (99.37 %), were determined. The kinetic non-isothermal degradation supplied the order of thermal reaction (0), the activation energy (96.14 kJ mol−1) and the frequency factor (3.130 × 10−7 min−1). The diffraction of X-rays presented well defined signs in the angles 5.5°, 16.3°, and 44.18° 2θ, suggesting crystalline structure. Scanning electronic microscopy exhibited needle morphology. LPSF/GQ-130 presented Type-III isotherm adsorption/desorption, with a superficial area of 81.3529 m2 g−1 and water content calculated at 1 % using the Karl Fisher method. Laser granulometry calculated its granulometry between 11.65 and 13.10 μm, thus it was characterized as a very fine powder. The prototype was classified as insoluble in water (<0.0187 μg mL−1) and soluble in acetone and acetonitrile, and exhibits instability in basic pH (100 %) and oxidative conditions (30–70 %). In thermal compatibility the excipients PVP K-30, Compritol® 888 ATO, and MYRJ® 59 seem to exercise a protective thermal activity for the prototype.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thiazolidinediones (TDZs) were first developed by Japan in 1980, in the midst of research around the thiazolidine ring, which presented great scientific interest due to its chemical properties and biological activities [1]. Due to its insulin-sensitizing action, TDZs were initially exploited for the treatment of type II diabetes [2, 3]; however, subsequent studies have detected the neuroprotective action of some TDZs against cerebral ischemia and intracerebral hemorrhage [4–7], suggesting, therefore, its anti-inflammatory activity.
Pharmacologically, these molecules act as agonists for peroxisomal proliferator-activated receptors of the gamma subtype (PPAR-γ), a transcription factor involved in metabolism and cellular regulation and differentiation [8]. Although originally believed that PPAR-γ would only influence in the differentiation of adipocytes and glucose homeostasis [9], its role, recently discovered, in the inflammatory response has been shown to be sufficiently important. According to Chung et al. [10], this receptor inhibits the expression of inflammatory genes, modulating, among other ways, the action of NF-kβ, AP-1, signal transducers, and activators of transcription. Additionally, besides its role as a transcription factor, the activation of PPAR-γ provokes a transrepression performance of macrophage inflammatory genes [11].
Thus, a great volume of preclinical studies have been published this decade exploring this activity, as pointed out in the systematic review carried out by White and Murphy [12]. In this context, the Laboratório de Planejamento e Síntese de Fármacos of the Universidade Federal de Pernambuco, Brazil, synthesized LPSF/GQ-130, TDZ that demonstrated, through in vitro studies in mice, inhibition of the inflammatory process in up to 75.1 % and low acute toxicity of the molecule [13], thus presenting itself as a powerful candidate for a medicine.
However, since it is a new chemical entity (NQE), the physical–chemical properties of LPSF/GQ-130 are practically unknown. Thus, before providing any technological intervention aiming at the rational description of the pharmaceutical formularization, its identification becomes vital, beginning with its physical–chemical characterization, besides studies that guide issues of stability [14], with the objective to establish a standard of molecule identification, guaranteeing its quality, safety, and effectiveness.
Aiming to achieve this objective, characterization studies of the prototype at the molecular, particle, and agglomerate levels were carried out. For certification of its chemical structure, nuclear magnetic resonance techniques (NMR) 1H and 13C, along with absorption spectrometry in the Fourier transform infrared (FTIR) and mass spectrometry (MS) were used. Preliminary stability studies of the NQE were carried out by means of forced degradation, in oxidative and hydrolytic conditions. In the ambit of particle thermal analysis techniques, X-ray diffraction (XRD) and scanning electron microscopy (SEM) were used. For agglomerate characterization, laser granulometry (LG) techniques and determination of the amount of sample humidity using the Karl Fischer method were used. Finally, NQE compatibility to different pharmaceutical excipients was carried out by thermogravimetry.
Materials and methods
LPSF/GQ-130
LPSF/GQ-130, 5-(4-chloro-benzidilene)-3-(2,4-diclorobenzil)-thiazolidine-2,4-dione, were granted by the Laboratório de Planejamento e Síntese de Fármacos (LPSF), situated in the Departamento de Antibióticos, Universidade Federal de Pernambuco.
Solvent and reagent
The solvents: Vetec® methyl alcohol P.A. and ethyl alcohol P.A. (lots 1007904 and 1002744, respectively), Dynamic® acetone P.A. and dichloromethane P.A., (lots 42061 and 1002744, respectively), Carlo Erba® acetonitrile (lot V9E853259E). The water used in the experiments was ultrapure (MILLI Q®). The solvents used for the preparation of the mobile phase and samples destined for the study of forced degradation were filtered in Millipore® filter membranes with a porosity of 0.22 μm and with 0.45 μm of cellulose. The substances: Vetec® sodium hydroxide, potassium phosphate dibasic, hydrochloric acid, and hydrogen peroxide.
Characterization of LPSF/GQ-130 raw material
Molecular characterization
Fourier transform infrared (FTIR)
The infrared spectra were obtained using PerkinElmer® (Spectrum 400) equipment with an attenuated total reflectance (ATR) device (Miracle ATR, Pike Technologies Spectroscopic Creativity) with a zinc selenide crystal. The samples to be analyzed were transferred directly to the ATR device compartment. The spectra were obtained with scans from 4,000 to 650 cm−1 in a resolution of 4 cm−1.
Nuclear magnetic resonance
The LPSF/GQ-130 samples were prepared in penicillin tubes and solubilized in deuterated chloroform, and were analyzed in a 400 MHz nuclear magnetic resonance device (Varian®).
Mass spectrometry
MS was carried out in a Shimadzu® mass spectrometer and its samples were prepared for direct infusion ESI (negative mode) MS analysis. The sample of LPSF/GQ-130 was first solubilized in acetonitrile (HPLC grade) and injected in small aliquots into the equipment. Acetonitrile, methanol, and water as a mobile phase were used; its retention time was 0.647 min.
Preliminary study of forced degradation
The previous studies of hydrolytic degradation were conducted with LPSF/GQ-130 raw material, using solutions of HCl, NaOH, ultrapure water, and HPLC grade acetonitrile for verification of hydrolysis acid, basic, and neutral, respectively, room temperature and with protection from light. For hydrolysis verification, the samples were solubilized in a 0.01 M solution of HCl and NaOH, 0.1 and 1 M of acetonitrile/water (60:40), evaluating behavior by HPLC after 72 h of exposure. Neutral hydrolysis was evaluated, varying only the time of exposure to the aqueous solvent, also after 72 h of exposure. Oxidative degradation was conducted using a hydrogen peroxide solution of 1, 3, and 9 % in a solution of acetonitrile/water (60:40) and evaluated for its behaviors for HPLC–DAD after the same time of exposure used for hydrolyses.
Since the prototype does not possess an indicative method of stability that determines the prototype contents and its related substances, we opted to use the conditions suited for rosiglitazone and pioglitazone, molecules structurally similar to LPSF/GQ-130, described in the works of Gomes [15] and Varanasi et al. [16]. Thus, the authors used column C18 reverse phase, with an acetonitrile/dihydrogen phosphate mobile phase in the ratios of 55:45 and 35:65 (v/v), respectively. The final pH of this mobile phase was adjusted to around 6.0. The analysis was carried out under a flow of 1.2 mL min−1 and an oven temperature of 25 °C. For verification of the degradation percentage, an LPSF/GQ-130 calibration curve in the concentrations of 25, 50, and 100 μg mL−1 in a solution of acetonitrile/water 60:40 were obtained, which showed r 2 = 0.999. The peaks observed in the HPLC had its pureness guaranteed by LC Solution 1.0 Shimadzu® software, in Shimadzu® equipment equipped with a quaternary system of LC-20ADVP pumps, degasifier Model DGU-20A, PDA detector model SPD-20AVP, oven model CTO-20ASVP, automatic injector model SIL-20ADVP, and controller model SCL-20AVP.
Particle characterization
Determination of the melting point and purity
The DSC curves for LPSF/GQ-130 were obtained by means of a Shimadzu® calorimeter, Model DSC-60 under a 50 mL min−1 atmosphere of nitrogen flow, the sample masses were analyzed at around 2 mg (±0.1), packed in aluminum hermetically-sealed sample holders. The DSC curves for thermal characterization and determination of the pureness of the prototype were obtained, respectively, at a heating rate of 10 and 2 °C min−1, until the temperature of 300 °C was reached, in triplicate. Before assay DSC calibration was carried out by means of the melting point of indium (156.6 ± 0.3 °C) and zinc (419.6 ± 0.3). The heat flux and enthalpy were calibrated by means of the melting point of indium (18.59 ± 0.3 J g−1), under the same conditions as the samples. The thermoanalytical data were analyzed by means of Shimadzu® TA-60WS® (Thermal Analysis) version 2.20 software.
Thermal degradation
TG/DTA curves as well as the first one derived from TG (DTG), were used for characterization of the prototype and analysis of the degradation kinetics by means of a Shimadzu® thermal balance, Model DTG-60H, in a nitrogen atmosphere with a flow of 50 mL min−1, the mass of the samples analyzed at around 3 mg (± 0.2) packed in aluminum sample holders. Characterization was carried out by heating the samples to a temperature range from 25 to 600 °C at the heating rate of 10 °C min−1. Before testing, instrument calibration, using a sample of monohydrate calcium oxalate, was verified with defined losses of mass, and for temperature calibration and melting enthalpies indium and zinc were used as a reference.
The non-isothermal kinetic study was carried out according to Ozawa’s method [17], heating samples up to 600 °C, and with heating rates (A) from 5, 10, 15, and 20 °C min−1. For data handling, the model proposed by Ozawa was used [17], utilizing the software previously described above. The angular coefficient (slope) of the graph that correlates log A versus 1/T (K−1) provides the process to E a. The values of the factor frequency (A) and order of reaction are also obtained in this kinetic study, from the graph that correlates the residual mass of the sample by the reduced time in minutes [18–20].
X-ray diffraction
The prototype diffractogram was obtained in a SIEMENS Diffractometer (X-ray diffractometer, D-5000) equipped with copper anode. The samples were analyzed at the interval angle 2°Θ of 2–60 to a digitalization speed of 0.02°2Θ s−1. The samples were prepared in glass supports with a fine layer of powdered material without any solvent.
Scanning electron microscopy
The samples were mounted on double-faced carbon ribbon, and metallized with gold for 15 min. The analyses were carried out on an FEI® microscope, model Quanta 200F, with 200 kV of acceleration voltage, low vacuum (0.5 Torr), FEG, and secondary electrons emitter as a way for image acquisition.
Determination of quantitative solubility in water
Excess LPSF/GQ-130 was placed in a test tube which was filled with 10 mL of MilliQ® water. The mixture was kept in a bain-marie with magnetic agitation and a temperature of 37 °C for 24 h. The samples, previously filtered in a Millipore filter®, were read according to the described method in the preliminary study of forced degradation. The analysis was carried out in quintuplicate. The method’s quantification limit was calculated according to Eq. 1.
Determination of the qualitative solubility in organic solvents
Seven milligrams of LPSF/GQ-130 were added to three test tubes, to which was added 1 mL of ethanol, acetonitrile, and acetone, with subsequent sonication lasting 5 min. The procedure was repeated until reaching the necessary amount of solvent for the attainment of a limpid solution. Solvents that were necessary in lesser amounts were linked to greater power of solubilization of the prototype [21]. Measurements were carried out using a UV-mini (Shimadzu®) spectrophotometer, using distilled water previously filtered through a Millipore filter® as a baseline.
Characterization of the agglomerates
Laser granulometric analysis
The experiment was carried out by wet means, using ethanol as a means of dispersion. The preparation of the LPSF/GQ-130 suspension in ethanol underwent agitation by ultrasound for a 5 min duration. The analysis was carried out in a particle-size distribution analyzer (Microtac®, S3500), with an SDC measurement module, 70 % flow rate and the Fraunhofer diffraction model.
Determination of superficial area and particle porosity
The measurement of specific superficial area and porosity was obtained by physical adsorption of nitrogen to powder material, by means of the Brunauer–Emmett–Teller (BET) method. Therefore, an analyzer of superficial area, Micrometrics® ASAP 2440, was used, equipped with software to determine the superficial area (S BET). Approximately 100 mg of the samples were weighed which were degasified for 48 h at 110 °C to remove any adsorbed material on the surface. The samples of the LPSF/GQ-130 prototype were analyzed with an atomization temperature of 160 °C in an air flow of 350 mL h−1.
Determination of the raw material humidity content
The determination of the moisture by the volumetric method was based on aquametry with Karl Fisher reagent, in Schott Titroline® alpha equipment. The moisture content was estimated by taking as a base the amount of CombiTitrant 5 reagent (Brand: Merck; Lot: HX083818), with its respective correction factor, necessary to titrate the water present in the aliquot of the assayed sample. Dry methanol was used as a solvent. The calculation of the moisture content was carried out using Eq. 2.
where v volume of the Karl Fisher reagent spent to titrate the water present in the aliquot of assayed sample; cf correction factor of the equivalent in water capable of being neutralized by the Karl Fisher reagent; AAS aliquot of the assayed sample; 100 calculation percentile factor.
Thermal compatibility
The study of the thermal compatibility of LPSF/GQ-130 to different pharmaceutical excipients was carried out in a Shimadzu® Thermal Analyzer, model DTG-60H, in a nitrogen atmosphere with a flow of 50 mL min−1, where the mass of the samples analyzed were around 3 mg (±0.2) and 6 mg (±0.2) for pure substances and their physical mixtures, respectively. All of the samples were packed in platinum sample holders and subjected to a 10 °C min−1 rate of heating, from 25 to 600 °C. For evaluation of the results the variation of the initial melting temperatures (T onsetF) and degradation (T onsetD) of the physical mixtures (PM) in relation to the pure LPSF/GQ-130 were taken into consideration. The PM were obtained at the ratio of 1:1 (w/w) with PVP K-30, Starch 1500, Tablettose, Neusilin FH2, PEG 6000, Compritol 888 ATO, MYRJ 59 (polyethylene glycol 100 stearate), and Polysorbate 80. The choice of excipients aimed at the attainment of solid dispersions and was based on the works of Chowdary et al. [22], Mishra et al. [23], and Amhed [24], who had obtained these systems for Pioglitazone.
Results and discussion
Molecular level characterization
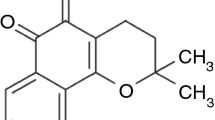
The LPSF/GQ-130 molecule (Fig. 1) was elucidated by means of FTIR, NMR of 13C and 1H, and MS. Beginning with the first technique the axial deformation band of the C=O (λ = 1,670 cm−1) bond is evident; the typical absorption bands of the C–N (λ = 1,108.86 and 1,085 cm−1) bond, added to the absence of bands in the typical region of vibration of the N–H (3,700–3,400 cm−1) bond, showing clearly the amide function; the bands of aromatical C=C (1,606 cm−1); besides bands in characteristic frequencies of benzene 1,4 disubstituted (λ = 813.28 cm−1); benzene 1,2,4 trisubstituted (λ = 929.12 cm−1); axial deformation of aryl chloride (λ = 1,146.21 cm−1); sulfonamide e (λ = 1,332.90 cm−1) (Fig. 1).

Infrared spectrum of LPSF/GQ-130
The spectrum obtained from the NMR1H showed vinylic hydrogen (7.871 ppm); aromatical hydrogens (agglomerated signs in chemical displacements between 7.153 and 5.455 ppm, which when added include an integral area of seven); and hydrogens bonded to Csp3 (5 ppm, with an integral area of two). In the NMR spectrum 13C, the presence of Csp3 (42.534 ppm); two bonds C=O (166.968 and 165.683 ppm); and aromatical carbons (relative to 12 signs between the chemical displacements from 129.636 to 136.932 ppm) can be confirmed. Presently, MS negative ionization (Fig. 2) allowed for the identification of deprotonated (M−1, m/z = 398.74) and decarboxylated (M−29, m/z = 369.93) LPSF/GQ-130.

Mass spectrometry of LPSF/GQ-130 obtained by direct infusion ESI (negative mode)
The preliminary study of forced degradation of LPSF/GQ-130 was carried out in conditions of hydrolysis and oxidation, since these are the most common due to the presence of oxygen and humidity in the atmosphere [25]. Thus, in acid hydrolysis (HCl 0.01, 0.1, and 1 M) there was degradation of around 10 % of the prototype in a 72-h exposure period, not demonstrating a relationship to the acid concentration, suggesting that the generated instability is due to the acid catalysis of the hydrolysis reaction, probably due to the displacement of the balance of dissociation of the water in the reaction.
On the other hand, in the same exposure period, the percentages of degradation shown in the basic conditions (NaOH 0.01, 0.1, and 1 M) demonstrated a well-known instability, since in all the concentrations of NaOH used; approximately 100 % of the prototype underwent degradation. The neutral condition did not present a significant degradation percentage (<10 %) in 72 h of analysis, where NQE was stable in these conditions.
Particle level characterization
The thermal profile of LPSF/GQ-130 crystals was determined from the sum of information obtained through DSC and DTG/DTA curves. Thus, the acute endothermic peak shown in the DSC curve (Fig. 3) in a temperature of 171.3–176.5 °C (–123.02 J g−1) can be related to its melting peak, since there was no event of mass loss in the TG/DTA curves (Fig. 4) at the same range of temperature. Additionally, the curves obtained demonstrate that the degradation of the prototype occurs in the range of 238.3–297.4 °C, with 98.6 % loss of mass (Δm). In this way, the temperature range can be identified in which the prototype shows itself thermally stable.

DSC curve of LPSF/GQ-130 under a 50 mL min−1 atmosphere of nitrogen flow and at a heating rate of 10 °C min−1

TG/DTG and DTA curves of LPSF/GQ-130 under a 50 mL min−1 atmosphere of nitrogen flow and at a heating rate of 10 °C min−1
According to Oliveira [26], it is possible to calculate the pureness of the samples from the linearization of the melting event observed in the DSC curve. This mathematical artifice is reached through the application of the Van’t Hoff (Eq. 1) equation, where it calculates the mole fraction of the material impurities, thus allowing for the indirect calculation of its purity. This method is very important, above all for drug prototypes because it does not need a model for its execution and interpretation [27].
This equation corresponds to a melting temperature of the impurities in the melting process (T s); the melting temperature of 100 % pure analyte (at Kelvin) (T o); the mole fraction of the impurities in the liquid phase (X 2); the melting heat of the pure sample (ΔH); and the gas constant R (8.3143 J K−1 mol−1).
For this analysis, Araújo [27] affirms that a heating range of up to 2 °C min−1, in virtue of the reports of scientific literature that point to the increase of the heating range as a critical point with respect to the reduction of the purity. This parameter is required because Van’t Hoff’s equation is only valid for balanced conditions, and therefore lower ranges are recommended. Thus, the purity of 99.37 ± 0.19 % and the impurities correlation factor of 11.13 % for the sample of LPSF/GQ-130 were obtained, the lot used was able to be considered as pure.
Since the TG curves presented a variation of mass, it was possible to calculate the kinetics of the non-isothermal thermal degradation of the LPSF/GQ-130 by applying the Ozawa model [28–30]. According to Soares-Sobrinho et al. [30], the knowledge about the physical–chemical properties and thermal drug and excipient decomposition comprises one of the requirements for development of new pharmaceutical products. Sovisi et al. [31] emphasize that, among determined parameters by this method, activation energy (E a) possesses the most interest to evaluate the stability of pharmaceutical compounds, able to be used for the establishment of a comparative profile among various drugs and their associations by means of the obtained E a values, the more activation energy, the more thermal stability of the compound.
Thus the method described by Ozawa, obtained by linear approximation based on integral calculus beginning with Arrhenius’ equation is very much used [17, 19, 20, 32–34]. As visualized in Fig. 4, the NQE presents thermal stability until approximately 238 °C and melts in the range of 171.3–176.5 °C, without alteration of mass. This verification is important in view of some substances, such as diclofenac sodium, which can undergo decomposition and/or cyclization before reaching the melting temperature depending on the heating rate and the atmosphere of the assay and, as a consequence, can have errors in the determination of its melting point [35].
In the assays of non-isothermal kinetic analysis with rates of heating (A) 5, 10, 15, and 20 °C min−1 TG curves were obtained that provided the correlation of log A versus 1 K−1(Fig. 5), from the model proposed by Ozawa [17].

a Heating rate logarithmic curves (log A) function of temperature (K−1); b graph of the function G(X) of the inverse temperature of LPSF/GQ-130
The data proceeding from TG curves make possible the determination of the E a (96.14 kJ mol−1) and of the factor frequency (3.130 × 10−7 min−1), beyond the order of the reaction of the thermal decomposition process of LPSF/GQ-130, calculated at zero. In accordance with the last result, the reaction of the loss or thermal decomposition of the prototype can be inferred independent of the reagent concentration and is constant with regard to the time [36]. All the data were obtained through the graph that correlates sample residual mass by the reduced time (Fig. 5b). The graph in question demonstrated a good correlation of the assays carried out under four heating rates.
Agglomerate level characterization
LG gave evidence of the average diameter of the agglomerate particle of the prototype at 12.48 μm. This technique was established as an international standard [37] for generating results comparable to those obtained by the method indicated in the Brazilian Pharmacopeia 5th edition [38]. Additionally, it was possible to determine, from the granulometric distribution curve (Fig. 6), the range of the average prototype granulometry at 11.65–13.10 μm, the result used as a parameter for future quality studies to be carried out on other lots of this material. Such results define this material as a fine powder, according to the Brazilian Pharmacopeia 5th edition.

Distribution curve and cumulative relative frequency of LPSF/GQ-130 agglomerates
According to Castellanos et al. [39], particles that possess a diameter below 30 μm present dominant interparticular cohesiveness. Such a phenomenon intensely compromises the raw material flow properties, and this represents a critical point in diverse unitary processes of production of pharmaceutical forms, such as mixture, compression, adequate filling of the matrices, and transposition of scale, among others [40, 41].
According to Torrado and Torrado [42], the knowledge of the crystalline form of the material in question is essential for the rational planning of the development of a pharmaceutical form, because the geometry in which crystals present themselves can signal how easily this can be formulated, also the ambit of the flow properties [42, 43].
According to Nery et al. [44], spherical particles present compression behavior and foreseeable flow for relatively simple models. Meanwhile, deviation from the spherical form or, difficult forms to be described by simple geometric figures, present difficulties in the forecast of its productive performance.
Thus, in accordance with the results obtained by scanning electronic microscopy and XRD, the crystalline behavior of the prototype can be verified, since the XRD presented well defined peaks in angles 5.5°, 16.3°, and 44.18° 2θ (Fig. 7), besides the absence of characteristic signals of an amorphous state, found in the 0°–10° region. However, it is possible that there exists only small amorphous fractions, in an inferior ratio of 1 % (Nery et al.) [44], given the equipment sensitivity. Additionally, micrographs emitted by the SEM (Fig. 7b) suggest needle geometry of the crystals. The crystalline structures presented adsorption/desorption isotherm type-III (Fig. 8), related to non-porous solids associated with mesopores (pores with a diameter of 20–500 Å) [45], as usually cited by the scientific literature. The superficial area was determined to be 81.3529 m2 g−1. The water content of the sample was around 1 %.

a Diffraction profile of LPSF/GQ-130; b micrograph crystals of LPSF/GQ-130 obtained by SEM

Absorption and desorption isotherm of LPSF/GQ-130
According to Saifee et al. [46], crystalline structures in the needle form present inadequate properties of flow when compared with particles of cubical or rounded morphology, where the intervention of excipients is necessary to submit them to diverse industrial processes destined for the attainment of medicines. Thus, the material is presented, a priori, inadequate for production of pharmaceutical forms, and the necessity of technological intervention of this material is evident, in order to avoid mixture heterogeneity and eventual problems of production.
The accurate quantification of the aqueous solubility of the prototype cannot be calculated, because it presents itself as inferior to the limit of quantification of the method used by HELC. Thus, it can be affirmed that this parameter assumes a value of <0.0187 μg mL−1. The aqueous insolubility of the NQE represents one of the most challenging aspects in the development of pharmaceutical forms administered orally [47]; and therefore, several techniques have been used to develop the hydrosolubility of pharmaceutical compounds, and thus to get profiles of adequate dissolution and biodisponibility [48]. According to Alves et al. [48], the attainment of solid dispersions represents a work of major industrial application because it deals with one simple low cost technique, besides supplying satisfactory results.
In the evaluation of qualitative solubility in organic solvents it was proven that LPSF/GQ-130 is more easily soluble in acetone and acetonitrile than in ethanol (Table 1). However, although acetone presents satisfactory parameters, its use is not diffused in molecules with similar chemical structure of the prototype in question, besides absorbing UV radiation near the classic wave length of organic molecules and rarely being used in high efficiency liquid chromatography with diode ray detection (HELC-DAD). Additionally, acetonitrile was used as much as a component of the samples as it was a mobile phase in diverse studies carried out with rosiglitazone, a structurally similar substance to LPSF/GQ-130 [15, 49]. For this reason, acetonitrile seems to be ideal as a solvent and/or co-solvent in the preparation of samples in solution of this prototype.
Thermal compatibility
Figures 9, 10, and 11 present, respectively, DTA, TG, and DTG curves of LPSF/GQ-130 and its PM. DTA curves demonstrate that all excipients provoke variations in the initial melting temperature of the prototype; however, the majority of them are insignificant variations. DTA curves of the PM with PVP K-30, PEG 6000, MYRJ 59, and Compritol 888 ATO demonstrate considerable anticipation of the melting of LPSF/GQ-130 (Table 2). This fact reflects that, in the presence of these excipients, the NQE crystals weaken; since, a lesser amount of energy is necessary to undo the crystalline network (melting), suggesting an interaction between substances involved [50]. On the other hand, to weaken the crystalline structure also reflects an increase of aqueous solubility of the substance, this being a beneficial consequence for LPSF/GQ-130 that presents a very low solubility in water. In accordance with the TG curve and its derivative (DTG) (Figs. 10, 11) it is possible to perceive that, although the anticipation of the initial melting temperature, there was retardation in the initial temperature of degradation for the excipients PVP K-30, MYRJ 59, and Compritol 888 ATO (Table 2). In this case, thermal stability was prolonged; since, the PM needs higher temperatures to initiate the process of thermal degradation of the prototype. This protection phenomenon of the active substance on the part of the polymer derives from the incorporation of the melted prototype in the polymer [31].

DTA curves of PM (1:1) under a 50 mL min−1 atmosphere of nitrogen flow and at a heating rate of 10 °C min−1

DTG curves of PM (1:1) under a 50 mL min−1 atmosphere of nitrogen flow and at a heating rate of 10 °C min−1

TG curves of PM under a 50 mL min−1 atmosphere of nitrogen flow and at a heating rate of 10 °C min−1
Additionally, the DTA curve demonstrates a significant reduction of the intensity of the melting peak of LPSF/GQ-130 in the PM with PEG 6000. This can be attributed to the partial solubilization of the prototype when the polymer is melted [51]. On the other hand, it is perceived that PEG 6000 kept the initial temperature of degradation of the prototype, where it was able to affirm that this excipient promotes an increase of solubility of the medicine, however it does not protect it thermally. The total solubilization is frequently observed in the diverse PM of other medicines with PVP K-30, and ibuprofene, naproxene, captopril, and cetoprofene [52]. However, for LPSF/GQ-130 the solubilization in melted PVP K-30 only seems to be partial, related to the saturation concentration of the prototype in a polymer, probably due to the difficulty of the formation of hydrogen bonds between substances, since both possess only acceptor points of hydrogen bonds, thus not possessing a donor for the establishment of the interaction.
Conclusions
The sum of information supplied by NMR of 1H, 13C, absorption spectrometry in the IR region and MS allowed for the identification of the molecule proposed by Santos et al. [13]. Thermal analysis proved very useful, because it was possible to determine its melting range, initial degradation temperature, thermal stability, prototype-excipient compatibility and, above all, the determination of its purity derived from Van’t Hoff’s equation, since the prototype does not yet possess an established standard. The set of physical–chemical techniques, such as granulometry, superficial area, and porosity, showed LPSF/GQ-130 properties as inadequate for medicinal development, where the necessity of technological intervention to bypass these problems has clearly become necessary. Finally, the preliminary study of forced degradation demonstrated the instability of LPSF/GQ-130 to basic and oxidative conditions (hydrolysis). The percentage of degradation through neutral hydrolysis was not significant.
References
Liesen AP, Aquino TM, Góes AJS, Lima JG, Faria AR, Alves AJ. Métodos de obtenção, reatividade e importância biológica de 4-tiazolidinonas. Quím Nova. 2008;31:369–76.
Buckingham RE. Thiazolidinediones: Pleiotropic drugs with potent anti-inflammatory properties for tissue protection. Hepatol Res. 2005. doi:10.1016/j.hepres.2005.09.027.
Barnett AH. Redefining the role of thiazolidinediones in the management of type 2 diabetes. Vasc Health Risk Manag. 2009;5:141–51.
Chen SD, Wu HY, Yang DI, Lee SY, Shaw FZ, Lin TK, Liou CW, Chuang YC. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem Biophys Res Commun. 2006;351:198–203.
Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by the peroxisomes proliferators-activated receptor-γ agonist rosiglitazone. J Neurochem. 2006;97:435–48.
Pereira MP, Hurtado O, Cárdenas A, Boscá L, Castillo J, Dávalos A, Vivancos J, Serena J, Lorenzo P, Lizasoian I, Moro MA. Rosiglitazone and 15-deoxy-∆12,14-prostaglandin J2cause potent neuroprotection after experimental stroke through noncompletely overlapping mechanisms. J Cereb Blood Flow Metab. 2006;26:218–29.
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor γ in microglia/macrophages. Ann neurol. 2007;61:352–62.
Michalik L, Auwerx J, Berger JP, et al. International union of pharmacology LXI peroxisome proliferator-activated receptors. Pharmacology. 2006;58:726–41.
Moraes LA, Piqueras L, Bishop-Bailey D. Peroxisome proliferator-activated receptors and inflammation. Pharm Ther. 2006;110:371–85.
Chung JH, Seo AY, Chung SW, Kim MK, Leeuwenburgh C, Yu BP, Chung HY. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res Rev. 2008;7:126–36.
Souza TRCL, Marques GS, Vieira ACQM, Freitas JCR. State of the art of anti-inflammatory drugs. Pharmacotherapy. 1st ed. Croácia: InTech; 2012.
White AT, Murphy AN. Administration of thiazolidinediones for neuroprotection in ischemic stroke: a pre-clinical systematic review. J Neurochem. 2010. doi:10.1111/j.1471-4159.2010.06999.
Santos LC, Uchôa FT, Moura RO, Lima MCA, Galdino SL, Pitta IR, Baerbe J. Synthesis and anti-inflammatory activity of new thiazolidine-2,4-diones, 4-thioxothiazolidinoes and 2-thioxoimidazolidinones. Heterocycl Commun. 2005;11:121–8.
Rodrigues OP, Cardoso TFM, Silva MAF, Matos JR. Aplicação de Técnicas Termoanalíticas na Caracterização, Determinação da Pureza e Cinética de Degradação da Zidovudina (AZT). Acta farm Bonaer. 2005;24:383–7.
Gomes P, Sipel J, Jablonski A, Steppe M. Determination of rosiglitazone in coated tablets by MEKC and HPLC methods. J Pharmaceut Biomed. 2004;36:909–13.
Veranasi VSKK, Veeraraghavan S, Potharaju S, Satheeshmanikandan TRS, Raghavan R, Swaroop KVVS. Validated high performance liquid chromatographic method for simultaneous determination of rosiglitazone, cilostazol, and 3,4-dehydro-cilostazol in rat plasma and its application to pharmacokinetics. Arzneimittelforschung. 2008. doi:10.1055/s-0031-1296509.
Ozawa TA. New method of analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38:1881–6.
Ozawa TA. Thermal analysis: review and prospect. Thermochim Acta. 2000;355:35–42.
Rodante F, Catalani G, Vecchio S. Kinetic analysis of single or multi-step decomposition processes-limits introduced by statistical analysis. J Therm Anal Calorim. 2002;68:689–713.
Cides LCS, Araújo AAS, Santos-Filho M, Matos JR. Thermal behaviour, compatibility study and decompositions kinetics of glimepiride under isothermal and nonisothermal conditions. J Therm Anal Calorim. 2006;84:441–5.
FARMACOPÉIA brasileira. 5.ed. Brasília: Anvisa, 2010. Part. 1, p. 125.
Chowdary KPR, Chandra DU, Mahesh N, Reddy TM, Gopaiah KV. Enhancement of dissolution rate and formulation development of pioglitazone – A BCS class II drug. J Pharm Res. 2011;4:3862–3.
Mishra SR, Ellaiah P, Jena PK, Nayak BS, Mishra G. An approach for enhancement of dissolution rate of pioglitazone HCl by solid dispersion. J Pharm Sci. 2011;2:2681–5.
Ahmed S, Ahmad Z, Baraskar N, Ahmed U, Siddiqui AR. Dissolution rate enhancement of pioglitazone hydrochloride by surface ternary solid dispersion. J Pharm Res. 2011;4:3606–8.
Carvalho JP, Santos AS, Sá AS, Teixeira CS, Nogueira MS. Estabilidade de medicamentos no âmbito da farmacovigilância. Fármacos e Medicamentos. 2008;102:22–7.
Oliveira MA, Yoshida MI, Gomes CL. Análise térmica aplicada a fármacos e formulações farmacêuticas na indústria farmacêutica. Quím Nova. 2011;34:1–7.
Araújo AAS, Bezerra MS, Storpirtis S, Matos JR. Determination of the melting temperature, heat of fusion, and purity analysis of different samples of zidovudine (AZT) using DSC. Braz J Pharm Sci. 2010;46:37–43.
Brown ME. Introduction to thermal analysis: techniques and applications. 2nd ed. Norwell: Kluer Academic Publishers; 2001.
Tiţa B, Marian E, Tiţa D, Vlase G, Doca N, Vlase T. Comparative kinetic study of decomposition of some diazepine derivatives under isothermal and non-isothermal conditions. J Therm Anal Calorim. 2008;94:447–52.
Soares-Sobrinho JL, Soares MFR, Lopes PQ, Correia LP, Souza FS, Macêdo RO, Rolim-Neto PJ. A preformulation study of a new medicine for chagas disease treatment: physicochemical characterization, thermal stability, and compatibility of benznidazole. AAPS Pharm Sci Tech. 2010;11:1391–6.
Sovizi MR. Investigation on decomposition kinetic of naproxen and celecoxib. J Therm Anal Calorim. 2010;102:285–9.
Cheng Y, Huang Y, Alexander K, Dollimore D. A thermal analysis study of methyl salicylate. Thermochim Acta. 2001;367:23–8.
Rodante F, Vecchio S, Catalani G, Tomassetti M. Application of TA and kinetic study to compatibility and stability problems in some commercial drugs. Remarks on statistical data. J Therm Anal Calorim. 2001;66:155–78.
Felix FS, Cides LCS, Angnes L, Matos JR. Thermal behavior study and decomposition kinetics of Salbutamol under isothermal and non-isothermal conditions. J Therm Anal Calorim. 2009;95:181–214.
Giordano F, Rossi A, Pasquali I, Bettini R, Frigo E, Gazzaniga A, Sangalli ME, Mileo V, Catinela SJ. Thermal degradation and melting point determination of diclofenac. Therm Anal Calorim. 2003;73:509–18.
Lachman L, Lieberman HA, Kanig JL. Teoria e prática na indústria farmacêutica. Lisboa: Fundação Calouste Gulbenkian; 2001.
International Organization for Standardization. ISO 13.320-1:1999: Particle size analysis laser diffraction methods—Part 1: general principles. Geneva, 1999.
Byung-man K, Lee JE, Jang-hyuk A, Tae-hong J. Laser diffraction particle sizing by wet dispersion method for spray-dried infant formula. J Food Eng. 2008;92:324–30.
Castellanos A, Valverde JM, Pérez AT, Ramos A, Watson PK. Flow regimes in fine cohesive powders. Phys Rev Lett. 1999;82:6–9.
Navaneethan CV, Missaghi S, Fassihi R. Application of powder rheometer to determine powder flow properties and lubrication efficiency of pharmaceutical particulate systems. AAPS Pharm Sci Tech. 2005;6:E398–404.
Sarraguça MC, Cruz AV, Soares SO, Amaral HR, Costa PC, Lopes JA. Determination of flow properties of pharmaceutical powders by near infrared spectroscopy. J Pharmaceut Biomed. 2010;52:484–92.
Torrado S, Torrado S. Characterization of physical state of mannitol after freezing-drying: effect of Acetylsalicylic acid as a second crystalline cosolute. Chem Pharm Bull. 2002;50:567–70.
Singhal D, Curatolo W. Drug polymorphism and dosage form design: a practical perspective. Adv Drug Del Rev. 2004;56:335–47.
Nery CGC, Arlete M, Pianetti GA, Vianna-Soares CD. Caracterização do fármaco hipoglicemiante glibenclamida. Rev Bras Cienc Farm. 2008;44:61–73.
Teixeira VG, Coutinho FMB, Gomes AS. Principais métodos de caracterização da porosidade de resinas à base de divinilbenzeno. Quím Nova. 2001;24:808–18.
Saifee M, Inamdar M, Dhamecha DL, Rathi AA. Drug polymorphism: a review. Int J Health Res. 2009;2:289–306.
Yu D-G, Yang J-M, Branford-White C, Lu P, Zhang L, Zhu L-M. Third generation solid dispersions of ferulic acid in electrospun composite nanofibers. Int J pharm. 2010. doi:10.1016/j.ijpharm.
Darlene L, Alves S, Andreza M, Lyra MD, Rolim LA, Maria G, Presmich A. Avanços, propriedades e aplicações de dispersões sólidas no desenvolvimento de formas farmacêuticas sólidas. Rev Ciênc Farm Básica Apl. 2012;33:17–25.
Venkatesh P, Harisudhan T, Choudhury H, Mullangi R, Nuggehally RS. Simultaneous estimation of six anti-diabetic drugs— glibenclamide, gliclazide, glipizide, pioglitazone, repaglinide and rosiglitazone: development of a novel HPLC method for use in the analysis of pharmaceutical formulations and its application to human plasma assay. Biomed Chromatogr. 2006;20:1043–8.
Stulzer HK, Rodrigues PO, Cardoso TM, Matos JSR, Silva MAS. Compatibility studies between captopril and pharmaceutical excipients used in tablets formulations. J Therm Anal Calorim. 2008;91:323–8.
Modi A, Tayade P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS Pharm. Sci. Tech. 2006;7:E87–92.
Tiţa B, Fuliaş A, Szabadai Z, Rusu G, Bandur G, Tiţa D. Compatibility study between ibuprofen and excipients in their physical mixtures. J Therm Anal Calorim. 2010;105:517–27.
Acknowledgements
Centro de Tecnologias Estratégicas do Nordeste (CETENE), Fundação de Ciência e Tecnologia do Estado de Pernambuco, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Medeiros Vieira, A.C.Q., Marques, G.S., de Melo, C.M. et al. Physical–chemical characterization of new anti-inflammatory agent (LPSF/GQ-130) and evaluation of its thermal compatibility with pharmaceutical excipients. J Therm Anal Calorim 115, 2339–2349 (2014). https://doi.org/10.1007/s10973-013-3358-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3358-y