
Abstract
Historical development of methods and theoretical basis of differential thermal analysis (DTA) are outlined. DTA is a procedure in which the heat transfer toward the sample plays an important function and the associated process in the sample is manifested by deviation of temperature difference from its background. This difference ΔT is not directly proportional to the rate of the process (dα/dt) but includes also the effect of heat inertia proportional to the slope dΔT/dt as it was derived and incorporated into DTA equation by Vold (Anal Chem 21:683–688, 1949), Borchardt and Daniels (J Am Chem Soc 79:41–46, 1957), and suggested to be corrected in the authors’ previous papers (1976). However, the correction with respect to heat inertia has so far been omitted (particularly after the boom of non-isothermal kinetics started by paper of Kissinger (Anal Chem 29:1702–1706, 1957)). DTA experiments with rectangular pulses realized by micro-heater inside the sample show that the correction of DTA signal employing calculated heat inertia term of the DTA equation is reliable but not yet fully sufficient. It was a reason to derive a more complete DTA equation including a term expressing the changes in the temperature field inside the sample during process. Possibilities for improving of DTA (and DSC) data processing are discussed. Itemized 150 references with titles.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
DTA: a historical introduction
Around 1880s the progress in metallurgy anticipated better investigations of thermal behavior of various iron-based alloys. The evaluation of heating and cooling curves (i.e., observation of stepwise temperature changes upon continuous heating) helped to reveal, e.g., the effects of carbon additions thus factually commencing thermal analysis as an important tool for a better elucidating of phase diagrams. The development of thermocouple, as an easy and accurate temperature measuring device, was employed by F. Osmond (1849–1912) and Roberts-Austen (1843–1902) [1, 2] in constructing devices to give a continuous record of the output from thermocouple (termed as a thermoelectric pyrometer). In 1899 Stanfield [3] published heating curves for gold, which almost stumbled upon the modern idea of differential thermal analysis (DTA, where the temperature difference between the sample and reference was recorded). The technique was afterward utilized by Kurnakov (1860–1941) [4, 5], who improved registration of his own instrument by the continuous photographic recording (DTA consequently utilized in [6–9]).
An initial theoretical inspection was given by Burgess (1874–1932) [10], who considered meaning of various curves in more details and concluded that the area of the inverse-rate curve is proportional to the quantity of heat generated divided by the rate of cooling. Correspondingly in 1909, there was elaborated a reliable procedure of preserving the high-temperature state of samples down to laboratory temperature by the procedure of sample quenching (i.e., freezing-in the high-temperature equilibrium state by fast cooling). It helped in the consistent construction of phase diagrams when used in the combination with optical microscopy (including metallography) used for the determination of mineralogical (phase) composition. Newly invented RTG analysis [11] (as introduced by 1912 experiment of M. von Laue (1879–1960) and by 1913 equation proposed by Sir W. L. Bragg (1890–1971) and W. H. Bragg (1862–1942)) showed that diffraction pattern can be used to determine crystal structure (and its parameters) and to identify solid phases (minerals). Both new techniques, DTA and XRD (X-ray diffraction), reveals in their resulting record characteristic singularities (or effects) called peaks. The processing of the records obtained by both methods has been based on evaluation of the position and the “size” (height, width or area) of peaks. It was obvious that some analogy on the peak evaluation could occur with some yet unclear consequences, which surprisingly has remained until today.
Important impact on the development of thermal measurements became the practical execution of DTA in the territory of former Czechoslovakia linked with the names O. Kallauner (1886–1972) and J. Matějka (1892–1960). They introduced thermal analysis as a novel technique during the period of the so called “rational analysis” of ceramic raw materials [12] particularly designed to investigate behavior of kaolinite [13] on heating. This material became a rather popular subject for an early DTA application [8, 14–18] particularly enhanced by R. C. Mackenzie (1920–2000) [20, 21] and his cooperation with the former Czechoslovakian scientists, initiating thus both the theoretical [22–24] and instrumental [25–27] consequences.
Only a small number of papers published in the period up to 1920 furnished some limited investigational details so that White [28] was first to show theoretically the desirable reproducibility using smaller samples describing thus more exhaustively the effect of experimental variables on the shape of heating curves as well as the apparent influence of temperature gradients and heat fluxes taking place within both the furnace and the samples. It was evident that DTA was primarily employed more as an empirical technique, although the experimentalists became generally aware of its quantitative potentialities. It even implicates the historical treaties by I. Newton (1642–1727) [29] the significance of which lies both in the range of temperature and in its instrumental presentation, particularly noting his famous “Law of Cooling”. However, the principal landmark in the theory of heat propagation was provided by J. B. J. Fourier (1768–1830) [30] who applied so called “Fourier series” to solve problems of heat transfer. In a system with a moving interface (e.g., melting or freezing), however, the heat transfer is connected with the so called “Stefan problem” [31] (named after Slovenian scientist J. Stefan [1835–1893]) indicated by G. Lamé (1795–1870) and B. P. Clapeyron (1799–1864) as early as in 1831 [32].
However, the early quantitative thermoanalytical studies were treated semi-empirically and based on an instinctive reasoning. Often mentioned is 1939 classical paper by Norton [14] on DTA execution in which he made rather excessive claims for its value both in the identification and in quantitative analysis (exemplifying clay mixtures [13–19]). Worth noting is the extensive work by Speil and colleagues [14, 15], who for the first time noticed the existence of heat inertia the appeal of which (subsequently Vold [33]) was overshadowed by inexhaustible troubles with failures caused by irreproducible experimental setups [19, 24, 28] (thermocouple positioning, sample geometry, etc.). The early DTA theory was also given in terms of specific heat changes by Sykes [34] in 1935. In 1951 Smyth [35] derived expressions describing theoretical heat flow through, and temperature gradients in, a DTA cylindrical sample. The literature of that period was extensively encompassed in [8, 9, 20, 23]. The first more comprehensive theories became accessible upon a series of papers [33–49] accentuating the early study by Soule [42] published in a less known French journal and mentioning also nearly unknown Czech-written papers [46–48]. The early theoretical books by Mackenzie [19], Berg [50, 51], and Piloyan [52] became important though the latter three were often overlooked due to their Russian language. More detailed historical roots are shown in Ref. [53–59] not forgetting early compendium by Mackenzie [9]. Sphere of modern thermal analysis emerged at the turn of seventies [60–65] together with a range of valuable books [66–73].
What is reflected by DTA curve?
Records of XRD and DTA measurements appear similar since they both represent curves consisting of peaks (characterized by their position and size) and backgrounds. In the case of the XRD the curve represents the intensity (I) of reflected X-rays as a function of the diffraction angle (θ). In the case of the DTA, the temperature difference ΔT as a function of temperature T (given by a temperature–time T–t heating regime curve) is displayed. However, this similarity is misleading, because they are substantially different methods. XRD measurement is non-destructive and does not change the state (structure and composition) of the sample under study and the diffraction peaks correspond to the reflections by atomic planes of a crystalline sample. DTA measurement is a method where the substance identity is altered and peaks on the DTA record detect changes on the state of the sample (having mostly irreversible character).
In the case of DTA the detected changes (due to processes the extent of which is expressed by the degree of conversion α) are associated with changes in enthalpy (ΔH) and the heat capacity of the sample (C P) which are manifested through the temperature deviation ΔT (=T S − T R)—the difference between the temperature (T S) of the sample and temperature (T R) of reference sample (reference sample should not exhibit any thermally detectable process during heating or cooling). This temperature difference (ΔT) is caused by the heat consumption (or production) during endothermic (or exothermic) transformation, but at the same time it is compensated by increased (or decreased) heat flux into the sample so that after completion of the conversion, the temperature difference is lowered to a level equal to the baseline. The DTA curves are reproducible only under identical condition of the same heating regime curve T R(t) usually in a linear form as T R = T 0 + Φ(t − t 0) where Φ is heating rate.
The resulting DTA curve is determined not only by the mechanism of transformation in sample but also by the rate of heat transfer between the sample and the surroundings. From a simple balance of heat fluxes for the sample and reference sample it follows [72, 73] that the measured temperature difference between the sample and the reference is not only proportional to the speed of transformation of the sample (dα/dt), but also the speed with which changes temperature difference (dΔT/dt). Under the condition of equality of heat capacities of the sample and the reference (including the sample holder) the temperature difference ΔT is given by
where t is time and K TΦ is so called apparatus constant (dependent on temperature T and on the externally applied heating rate Φ = dT/dt). In the case of heat-flux DSC, the measurement output is pre-calibrated as a difference between heat fluxes
so that the Eq. (1) has a form:
The term C P·(dΔT/dt) (or better C P·d(Δ(dq/dt)HF)/dt is a mathematical expression of the below-discussed heat inertia.
The practice and basis of DTA has been treated numerously [14–20, 33–52] implying the control of heat flux from the surrounding heaters where heat itself is assumed as a kind of physico-chemical reagent [55, 72, 73], which, however, could not be directly measured but merely calculated on the basis of the measurable temperature responses. We should keep in mind that the subsequently invented power-compensation DSC is of a different nature because it evaluates the authentic compensating heat fluxes instead of mere temperature differences where the heat inertia term is naturally absent [72, 73]. Particularly, the true DSC is monitoring the difference between the counterweighing heat fluxes by two extra micro-heaters, respectively attached to both the sample and reference to keep their temperature difference smallest (ΔT ⇒ 0), while the samples environment is maintained in the pre-selected (outer) temperature program. Only at this arrangement the corresponding equation did not contain the heat inertia term:
This technique was originally introduced by Eyraud and O’Neill in 1950s [74, 75] bearing thus a quite different measuring principle when comparing with the standard (already known) DTA because the temperature difference is not used for the entire examination but exclusively serves for the regulation, only [72–75].
On correct theoretical basis of DTA measurements
Let us repeat that the heat inertia term was quite overlooked in the early publications, although theoretical basis of differential measurement concentrated on questions associated with heat transfer [14–16, 33–52, 61–71]. In 1939 the first notice of heat inertia term appeared in the reports by Speil and colleagues [15, 16] and in 1949 in the work by Vold [33] (so far receiving 155 citations by WOS). We can repeat below her Eq. (8) from [33]:
where Δ0 T means a level of signal background, C S is heat capacity of sample and A ≈ K/C S. The equation (which is nearly equivalent to the Eq. (1) in the preceding paragraph) was obtained by Vold [33] via a complicated derivation which is starting with a typing error (bold) in dH/dt in place of dH/dT in the original equation dH/dt = (dH/dt)(dT/dt). It could be a reason why the equation was not very credible for citation of a majority of thermoanalysts. The results of Vold was detailed by Proks [46], who combined them with the results of other authors to derive the dependences of peak area and of position and height of extreme deviation (on the DTA curve) upon heating rate.
Nevertheless even well-written books on DTA [19, 24, 50–52, 66–71] did not persuaded this heat inertia design into a more widely accepted understanding because the influence of heat inertia on the whole peak area is realistically negligible so that it has practically no impact on the enthalpy measurements. This negligibility is due to the fact that the contribution of heat inertia at the onset stage of DTA peak is nearly compensated by the reversed contribution at the attenuation (run-down) stage of the peak. The effect of thermal inertia is actually represented by s-shaped curve and its addition to original DTA does not change the value of peak area. Also for the determination of various thermal criteria (such a glass-forming coefficients) this inertia term possesses negligible effect [76–78].
In 1957 the mentioned inertia term was used by Borchardt and Daniels [39] (824 WOS citations [79]), who applied DTA to study kinetics of homogeneous reactions in well-stirred liquid samples. They start with the balance equation (see their Eq. (3) in [39])
from which the Eq. (1) in preceding paragraph can be obtained substituting dH = ΔHdα and then dividing their equation by dt. The correction with respect to heat inertia is included also in their Eq. (13) [34] expressing the rate constant k of a homogeneous reaction
(where A is total area of peak and a is a part of this area for time interval t − t 0) and this correction is included in other further equations (from (12) to (15) in [39]). However, the heat inertia term was then neglected in the original [39] Eqs. (17), (19), (21), and (22) in accordance with the authors’ argument [39]: “… the quantities (C P dΔT/dt) and C PΔT are usually an order of magnitude smaller than the quantities to which they are added and subtracted. The results [39] show, however, that term „C PdΔT/dt varies from 0.634… to −2.70“ while the term KΔT “varies from 4.67 to 13.1 going through a maximum of 28.1”. For that reason the above neglecting seems to us to be incorrect because the heat inertia term has a significant influence (being asymmetrical on the level approaching the curve inflection points—differing at least 10 % from the original signal). The shape of kinetic curve and the derived kinetic parameters are extremely sensitive to this heat inertia consequence.
Blumberg [41] (49 WOS citations) applied DTA to study kinetics of heterogeneous reaction of silica with HF in a linearly heated bath. He used also a theoretical background with a thermal inertia term as it follows, e.g., from his Eq. (8) (in [41]). Nevertheless, at the same time Kissinger [80] proposed a kinetic evaluation based on the shift of the DTA peak apexes along with increased heating rates. Despite becoming one of the most quoted kinetic appraisals (with as many as 4,500 WOS citations [79]) it did not account on the prospective of heat inertia distorting effect nor did it affect its consequent mathematical endorsements [81]. Ten years later it was criticized and the characteristic temperatures were substituted by reaction rates derived from the slope of mounting part of a DTA peak by Piloyan et al. [82] (365 WOS citations [79]), however, missing the heat inertia again [52].
A first more adequate analysis was brought by Gray [83, 84], however, not yet elaborated in contemporaneous publications [60–71]. However, the detailed evaluation waited until the late 1970s in the comprehensive studies by Holba and colleagues [85–91] (published, unfortunately, in lesser known journals) piloting thus the full-size DTA equation in the form:
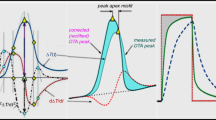
where ΔT DTA is the difference between temperature of proper sample (T S) and that of reference sample (T R), T W is the temperature of furnace wall, ΔK is the difference between coefficients of heat transfer between the furnace and sample holder (K S) and between the furnace and reference holder (K R), Φ = dT R/dt is the externally applied linear heating rate, K is the (in J K−1 s−1) so called “apparatus constant” of a given DTA instrument (depending on temperature T R and heating rate Φ), ∆ t H is the integral enthalpy change due to transition/reaction inside sample and α is the extent of transition (conversion). The recalculation of experimental DTA curve according to Eq. (2) gives an illustrative peak rectification graphically shown in Fig. 1.

Graphical representation of individual contributions composing a DTA peak assuming the standard DTA setup with samples (T S and T R) heat from the outside (T W), left insertion. Customary evaluation is based on the circumference bounded by the dashed as-measured DTA peak against dotted interpolated peak background. The true s-shaped peak background (solid line) due to the heat inertia does not practically contribute the peak resultant area, but the shape of DTA peak is seriously changed. The stepwise partitioning of peak area, necessary for determining the gradual progress of degree of reaction became different from that realized on basis of simply linearly interpolated peak background
Even though the final software proposal appeared in a respected journal of Thermochimica Acta [92] the perceptible effect of thermal inertia has not been accredited until today [93, 94] with few exceptions [95, 96] not mentioning the detailed description in the books by Šesták [73, 97, 98].
As a matter of curiosity, the pulse method (of flush heating) [99, 100] can be accentuated when originating from similar heat phenomena associated with deferred heat propagation, which is used to determine the entire heat diffusivity of studied samples [99, 100] later used in our model calculations [101, 102].
Another experimental arrangement was introduced by Svoboda and Šesták [103, 104] to simulate heat evolution in a DTA sample. Practically it was realized by inserting rectangular (electrically initiated heat) pulses [101, 102] into the readjusted DTA cell (containing an inserted micro-heater). This arrangement, however, reminded almost without public response though it well-approved the equation used for peak rectification, see Fig. 2, notwithstanding that such a pulse calibration is not unique [100].

Authorization of the DTA equation validity when containing the term of heat inertia (dΔT/dt) by reconstructing an inserted rectangular heat pulse. Resultant (DTA-like) peak (dashed line) becomes the instrumental response to the artificial rectangular input (dotted) due to the heat transfer. Curve refinement upon the application of DTA equation (particularly incorporating the term for heat inertia) yields rectified DTA peak (solid line). The greatest differences between the originally introduced and reconstructed heat process is in the regions near to the onset (start) and termination of rectangular pulse, where the heat flux (q) is abruptly changed (\( \ddot{q} \) ≡ d2 q/dt 2 → ±∞), see the upper right inserted scheme. This misfit can be explained as a consequence of abrupt changes in temperature field inside the sample and described by an additional correction term in DTA equation (see Eq. 9 as derived in [101]) using the rate dθ SM/dt, where θ SM is the difference between the surface-measured temperature and the temperature averaged over the whole sample volume
Factually, such appropriately made analysis shown in Fig. 2, confirms that the extent of DTA peak rectification is soundly established. The correct DTA equation [85–92, 97, 98] makes available a well-rectified curve (solid line), the shape of which is more similar to the original pulse (dotted), the differences still remaining near the onset (start) and the termination (end) of the heat pulse (Fig. 2, upper right scheme) exhibiting yet a certain degree of misfit. Therefore, the question arose what is the cause of this remaining insufficiency. The most plausible origin of such an incomplete rectification stays a simplification used in the formulation of heat-flux balances, where only single temperatures (T S and T R) are used to characterize temperature states of both samples. As a matter of fact, the temperature of any material body exposed to a definite heating and/or cooling is not uniform (even if the material is thermally inert not exhibiting any heat consuming and/or generating processes) so that their so called thermoscopic state of the real body should be described by a temperature field T(x, y, z), where x, y, z are sample space coordinates. The simplest description of temperature field can be used when a sample has a form of infinite cylinder where local temperature T r = T(r) is function of only one space variable—radius r. The estimation of this time-dependent non-uniform temperature field and its impact on the shape of DTA curve represented the main objective of our recent book chapter [101].
In the mentioned chapter [101], the equation for DTA curve is derived for a system of two infinite cylinder holders (one of which filled by the sample and the other one by the inert reference material) and proposed in the following form
where ΔC Φ ≡ r 2E (C RM − C SM), C Δ ≡ (r 2H − r 2E )C H + r 2E C SM, C θ ≡ r 2E C SM, θ SM ≡ T Sø − T EHS, T Sø ≡ (2/r 2E )·∫ rE0 (dT S(r)/dt)rdr and r E is the internal radius of holders, C RM and C SM is the heat capacity of sample (S) and reference (R) materials, θ SM is the difference between temperature averaged over whole volume of sample material and temperature measured on external wall of sample holder, r H is the external radius of holders, α G is the “global” extent of conversion (transition/reaction) averaged over whole volume of sample.
It is clear that temperature gradients, which naturally occur in every bulk sample [35, 105] are the source of rather deep dissensions often ignored or better disregarded in the recent state of thermal analysis. The topical development of DTA instrumentation has concentrated on an achievement of very precise outer surface temperature detection, going down to sensitivity of tenth or even hundredths of degrees neglecting, however, inhomogeneous and variable temperature field inside the body. It seems that thermoanalysts believe that a mere replacement of thermocouples (in DTA) by thermocouple batteries (in heat-flux DSC) or by highly sensitive electronic chips (in nano-calorimetry [106, 107]) moreover renaming DTA principle to DSC is a sufficient solution. Many DTA-resembling methods are described solitary on basis of difference of heat flows, Δ(dq/dt), between the samples, which factually substitutes the instrumentally observed (and eventually averaged) temperature difference according to Eq. (2) (Δ(dq/dt) ≈ KΔT). Such an approach [66–71, 93, 94] actually obscures the inherent nature of the as-received heat-flow DSC signal.
The resulting DTA curves are regularly subjected to a kind of signal leveling and data smoothing [108–114] which yields the so called desmearing procedures, which may be of help when furnishing the truer heat capacity–temperature function of the sample under study. Desmearing is actually filtering of the measured signal, which in turn was taken over from the XRD practice again [115, 116] and implemented to DTA/DSC practice [112, 113], which has nothing to do with the elimination of the effect of heat inertia by the peak rectification. Inherent practice of electric calibration of calorimetric measurements by heat pulses has been found functional; however, the consequent analysis of the resulting curve profiles is plausible and indispensable.
It is a matter of time when the rectification DTA and heat-flux DSC curves using terms correcting both the heat inertia effect (term proportional to dΔT DTA/dt) [66–71, 93, 94] and the effect of varying temperature gradient inside sample (the term proportional to the rate dθ SM/dt of the difference θ SM between the measured (outer) temperature of sample holder T EHS and the temperature T Sø averaged over whole volume of the sample [101, 102]) will be introduced to both the private and commercial practice of instrumentally available software. First encouraging signals can be seen in the former discrimination between the compensation and the heat-flow DSC by Illekova et al. [96] (who properly introduced for the latter method the term of heat inertia) and a current introduction of averaged sample temperature by Lyon et al. [117].
Perspectives in an advanced DTA assessment
It seems perceptible that the most essential impact of the above analysis [66–71, 93, 94] can be expected in the sphere of thermoanalytical kinetics based on the determination of the instantaneous degree of conversion of a process, α (or the rate of conversion dα/dt) [118–127] derived from the location of DTA peak apex [80, 81]. Such kinetics often involves the participation of DTA peak area which fractioning is not the same for the corrected and the uncorrected peak. The peak rectification [92] is well thought-out in the last two papers only [126, 127].
Already in the early seventies [85] we noticed an unusually strong correlation between the DTA peak areas and activation energies, E A, calculated from DTA curves representing the tetragonal-to-cubic transitions of the solid solutions Mn2CrO4–Mn3O4 (i.e., Mn2+xCr1−xO4) [72, 85, 118, 128], cf Table 1. Rough decoding of an apparent correspondence between the reaction rate and peak shapes led us to a factually deeper analysis of the heat process occurring under conditions of a DTA experimentation mounting the above-mentioned DTA equation [86, 87, 92].
Moreover, the kinetic analysis arising from procedure [92, 97, 98] shows a more deeper impact and evaluation complexity particularly touching discrimination of reaction mechanism [118–127] and associated activation energies [129, 130] of solid samples, where no instantaneous homogenization (e.g., by stirring as applied in [38]) can be employed to maintain the even sample temperature distribution during the studied reaction. This problem which could be called a dilemma of solid-state kinetics [97, 98] was not included neither identified in the earlier [52, 60, 66] nor in recent [118–125] papers. Beside early notices on thermal gradients [35], there subsists no appropriate attention with few exemptions [117, 118, 126, 127]. It is surprising that even very current contribution [125] does not touch such an obvious intricacy of thermal measurements and that even ICTAC kinetic committee keeps away from inserting the problem of thermal inertia and thermal gradients among their agenda [131, 132], though it has been recognized for long [91] and its inevitability indicated [127].
Heat transport problems are traditionally overwhelmed by difficulties arising from (often irreproducible) experimental setups and [66–71] and mathematical variability of evaluation methods [131, 132]. Credibility of derived kinetic data thus suffers drowning out the errors due to mathematical procedures [97, 98] and/or kinetic misinterpretation of consequential numbers [129, 130] always providing records capable for publication. In our earliest computer evaluation for BaCO3 transformation [92], we found that incorporation of heat inertia reduced the value of activation energy to about one half while not appreciably altering its reaction mechanism. On the other hand above discussed paper by Borchardt and Daniels [39] misguidedly simplified the Eq. (7) by neglecting the derivative term dΔT/dt due to its audible smallness, which could negatively affect kinetic end-users (see above arguments). Certainly a conflict can be awaited with respect of various computer programs [133–135] where such an extra procedure ought to be incorporated to adjust yet classical peak partitioning with an advanced subset due the heat inertia effect [91, 92].
Temperature is accurately monitored only over the sample surface, while any inner layers display different values. It is curious that such an apparent contradiction did not shift emphasis toward an improved instrumental judgment to furnish at least a mean sample temperature (e.g., mathematically averaged over the whole sample volume). Most of needed data are experimentally identifiable and sophisticated assets for a computer evaluation equally accessible. We are sorry to strongly voice that such existing negligence of the influence of real experimental conditions including thermal inertia and non-uniform temperature field indicate that the thermoanalysts prefer effortless data processing provided by instrumental producers before the legitimate respect to the physical reality of thermoanalytical experiments.
Even in the sphere of thermodynamics the deeper analysis of the s-shaped peak background (cf. Fig. 1) may provide a focal tool for improving data accuracy on heat capacities. It may even affect the method truthfulness when the thermal study is carried out under the temperature modulated program [136, 137], where the averaged (overdue) bulk temperatures behave differently than the surface (actually measured) temperature [101, 102].
A particular attention should be paid to the modified thermophysical procedure of the rate-controlled mode of thermal analysis [138], where the sample temperature is not adjusted by a constant external heating but only monitored as a variable for maintaining a constant reaction rate thus not suffering from such extreme gradients. Worth noticing are the special trends of nano-focused calorimetry [142, 143] because the upcoming prospect of thermal analysis may even go down to the nano- and quantum world [144] taking care of a very specific behavior of nano-composed and nano-assembled samples [145–150]. Measurement of such extreme setups and under so radical conditions brings extra difficulties often associated with the sample constrained states. At the same time, it will include a non-equilibrating side-line effect or competition between the properties of the nano-sample bulk and its entire surface (particularly when exposed to the contact with the cell [106, 107]). Increasing instrumental sophistication and sensitivity will provide possibility to look at the sample micro-/nano-locality [139–141] giving a better chance to search more thoroughly toward the significance of baselines, which contains additional but hidden information on material structure and properties (inhomogeneities, local non-stoichiometry, interfaces between order–disorder zones). It may even intervene with quantum measurements [144] if the sample thickens (often below μm) would interfere with the thermal vibration modes distorting thus the classical expression of thermal capacity [145] (e.g., stationary vs. dynamic).
Conclusions
There is a long lasting question why the current assessment of DTA peaks does not comprise heat inertia and temperature gradients even if those exist on every occasion of real measurements [35, 46, 73, 95]. The heat transfer analyses [97, 98, 101, 102] presented above (cf. Figs. 1, 2) definitely confirm not only the inevitability of insertion of the earlier suggested rectification term based on the heat inertia (dΔT DTA/dt) but lead also to the requirement for introducing an additional correction term respecting the changes in temperature field inside the sample dθ SM/dt [74] (where θ SM is the difference between the surface-measured temperature and the temperature averaged over the whole volume of sample). This situation always occurs during the temperature changes as a consequence of external heating or thermally assigned reactions and the authors anticipate an auspicious beginning of wide-ranging discussion in our websites Footnote 1 (see footnote) where other relevant papers are presented. On the other hand, the heat inertia term is zero for a compensation DSC device (by Perkin-Elmer) but it always survives a permanent part of the description of heat-flux DSC. The heat inertia term at a heat-flux DSC (as well as at DTA) cannot be reduced by diminishing the sample size [101, 102]. The effect of changes of temperature field in the sample cannot be instrumentally eliminated by a construction sophistication of any DTA and/or DSC apparatus.
Notes
See our open-access websites: http://www.thermotics.eu/and http://www.fzu.cz/~sestak.
References
Roberts-Austen WC. Fifth report to the Alloys Research Committee. Nature. 1899;59:566–7.
Roberts-Austen WC. Report 5. Proc Inst Mech Eng. 1899;35–102.
Stanfield A. On some improvements in the Roberts-Austen recording pyrometer, with notes on thermo-electric pyrometry. Philos Mag. 1898;46:59–82.
Kurnakov NS. Eine neue Form des Registrierpyrometers. Z Anorg Chem. 1904;42:184–202.
Kurnakov NS. Studies in the field of metallurgy. Moskva: Gos Nauch Techn Izd. 1954. p. 104 (in Russian).
Gibson RE. Pressure effect on α–β transition in quartz. J Phys Chem. 1928;32:1197–202.
Kracek FC. Polymorphism of sodium sulfate by thermal analysis. J Phys Chem. 1929;33:1281–303.
Rowland RA. Differential thermal analysis of clays and carbonates. Clays Clay Technol. 1954;169:151–64 (with 200 selected references on DTA compiled by Sans JF, pp. 159–63).
Mackenzie RC. Origin and development of DTA. In: Mackenzie RC, editor. History of thermal analysis, special issue of Thermochim. Acta, vol 73. Amsterdam: Elsevier; 1984. pp. 307–67 (including 250 DTA-sourced references, pp. 361–7).
Burgess GK. Methods of obtaining cooling curves. Bull Bur Stand. 1908–1909;5:199–225.
Hauptman HA. History of X-ray crystallography. Struct Chem. 1990;1:617–20.
Kallauner O, Matějka J. Beitrag zu der rationellen Analyse. Sprechsaal. 1914;47:423.
Matějka J. Chemical changes of kaolinite on firing; Chem Listy. 1919;13:164–6, 182–5 (in Czech).
Norton FH. Critical study of the differential thermal methods for the identification of the clay minerals. J Am Ceram Soc. 1939;22:54–61.
Speil S. Application of thermal analysis to clays and other aluminous minerals. US Bureau of Mines, Technical Paper 1944; R.I. 3764. p. 1–36.
Speil S, Berkenhamer LH, Pask JA, Davies B. Differential thermal analysis, theory and its application to clays and other aluminous minerals. US Bureau of Mines, Technical Paper 1945. p. 664–745.
Murray P, White J. Kinetics of the thermal dehydration of clays. Trans Br Ceram Soc. 1949;48:187–206.
Mackenzie RC. Modern methods for studying clays. Agrochimica. 1957;1:305–7.
Mackenzie RC, editor. The differential thermal investigation of clays. London: Mineral Society; 1957.
Mackenzie RC, Mitchell BD. Differential thermal analysis, a review. Analyst. 1962;1035:420–34.
Lombardi G, Šesták J. Ten years since Robert C. Mackenzie’s death: a tribute to the ICTA founder. J Therm Anal Calorim. 2011;105:783–91.
Šatava V. Differential thermal analysis. Silikáty (Prague). 1957;1:207–10 (in Czech).
Šatava V. Documentation on thermal analysis. Silikáty (Prague). 1957;1:240–52 (in Czech).
Eliáš M, Šťovík M, Zahradník L. Differential thermal analysis. Praha: Publ. House AVČR; 1957 (in Czech).
Mitchell BD, Mackenzie RC. An apparatus for DTA under controlled atmosphere conditions. Clay Miner Bull. 1959;4:31–4.
Šatava V, Trousil Z. Simple construction of apparatuses for automatic DTA. Silikáty (Prague). 1960;4:272–7 (in Czech).
Šesták J, Burda E, Holba P, Bergstein A. An apparatus for DTA in vacuum and regulated atmospheres. Chem Listy (Prague). 1969;63:785–9 (in Czech).
White WP. Melting point determination. Am J Sci. 1909;28:453–73.
Newton I. Scale graduum Caloris. Calorum Descriptiones & Signa. Philos Trans. 1701;22:824–9.
Fourier JB. Théorie analytique de la chaleur. Paris: Firmin Didot; 1822.
Stefan J. Über einige Probleme der Theorie der Wärmeleitung. Sitzungsber. Wiener Akad Math Naturwiss Abt. 2A, 1889;98:473–84.
Lamé G, Clapeyron BP. Mémoire sur la solidification par refroidissement d’un globe liquide Ann. Chim Phys. 1831;47:250–6.
Vold MJ. Differential thermal analysis. Anal Chem. 1949;21:683–8.
Sykes C. Methods for investigating thermal changes occurring during transformations in solids. Proc R Soc. 1935;148A:422–9.
Smyth HT. Temperature distribution during mineral inversion and its significance in DTA. J Am Ceram Soc. 1951;34:221–4.
Boersma SL. A theory of DTA and new methods of measurement and interpretation. J Am Ceram Soc. 1955;38:281–4.
Pask JA, Warner MF. Differential thermal analysis: methods and techniques. Bull Am Ceram Soc. 1954;33:168–75.
Borchardt HJ. Differential thermal analysis. J Chem Educ. 1956;33:103–9.
Borchardt HJ, Daniels F. The application of DTA to the study of reaction kinetics. J Am Chem Soc. 1957;79:41–6.
Borchard HJ. Initial reaction rates by DTA. J Inorg Nucl Chem. 1960;12:252–4.
Blumberg AA. DTA and heterogeneous kinetics: the reactions of vitreous silica with HF. J Phys Chem. 1959;63:1129–32.
Soule J. L’interpretation quantitative analyse der l’thermique differentiale. J Phys Radium. 1952;13:516–20.
Nagasawa K. DTA studies on the high-low conversion of vein quartz in Japan. JES Nagoya Univ. 1953;1:156–76.
Sturm E. Quantitative DTA by controlled heating rates. J Phys Chem. 1961;65:1935–37.
Speros DM, Woodhouse RL. Quantitative DTA: heats and rates of solid–liquid transitions. Nature. 1963;197:1261–3.
Proks I. Influence of temperature increase rate on the quantities important for evaluation DTA curves. Silikáty (Prague). 1961;1:114–21 (in Czech).
Šesták J. Temperature affecting the kinetic data accuracy obtained by TA measurements under constant heating rate. Silikáty (Prague). 1963;7:125–31 (in Czech).
Proks I. Effect of quantities controlling DTA on the difference between measured and theoretical temperatures. Silikáty (Prague). 1970;14:287 (in Czech).
Garn PD. Thermal analysis—a critique. Anal Chem. 1961;33:1247–55.
Berg LG. Rapid quantitative phase analysis. Moscow: Akad Nauk; 1952 (in Russian).
Berg LG. Introduction to thermography. Moscow: Akad Nauk; 1961 (in Russian).
Piloyan GO. Introduction to the theory of thermal analysis. Moskva: Izd Nauka. 1964 (in Russian).
Šesták J, Mareš JJ. From caloric to statmograph and polarography. J Therm Anal Calorim. 2007;88:763–68.
Šesták J. Some historical features focused back to the process of European education revealing some important scientists, roots of thermal analysis and the origin of glass research. In: Šesták J, Holeček M, Málek J, editors. Some thermodynamic, structural and behavioural aspects of materials accentuating non-crystalline states. OPS-ZCU Pilsen; 2011. p. 29–58 (with 166 useful references). ISBN 978-80-87269-20-6.
Šesták J, Hubík P, Mareš JJ. Historical roots and development of thermal analysis and calorimetry. In: Šesták J, Mareš JJ, Hubík P, editors. Glassy, amorphous and nano-crystalline materials. Berlin: Springer; 2011. p. 347–70 (with 125 useful references). ISBN 878-90-481-2881-5.
Šesták J. Phenomenology of non-isothermal glass-formation and crystallization. In: Chvoj Z, Šesták J, Tříska A, editors. Kinetic Phase Diagrams: nonequilibrium phase transitions. Amsterdam: Elsevier; 1991. pp. 169–276 (with 270 useful references). ISBN 0-444-88513-7.
Proks I. Evaluation of the knowledge of phase equilibria. In: Chvoj Z, Šesták J, Tříska A, editors. Kinetic Phase Diagrams: nonequilibrium phase transitions. Amsterdam: Elsevier; 1991. pp. 1–54 (with 170 useful references). ISBN 0-444-88513-7.
Proks I. The Whole is Simpler than its Parts: chapters from the history of exact sciences. Bratislava: Veda (Slovak Academy of Sciences); 2012 (in Slovak). ISBN 978-80-334-1158-5.
Holba P, Šesták J. Czechoslovak footprints in the development of methods of thermometry, calorimetry and thermal analysis. Ceram Silikaty. 2012;56:159–67 (with 230 useful references).
Murphy CB. Thermal analysis: state-of-art. Anal Chem. 1979;42:268R–76R.
Faktor MM, Hanks R. Quantitative application of dynamic differential calorimetry. Part 1.—theoretical and experimental evaluation. Trans Faraday Soc. 1967;63:1122–9.
Faktor MM, Hanks R. Part 2.—heats of formation of the group 3A arsenides. Trans Faraday Soc. 1967;63:1130–5.
Berg LG, Egunov VP. Quantitative thermal analysis I: mathematical problems of quantitative DTA. J Therm Anal. 1969;1:5–13.
Berg LG, Egunov VP. Quantitative thermal analysis III: effect of experimental factors in quantitative DTA. J Therm Anal. 1970;2:53–64.
Šesták J, Berggren G. Use of DTA for enthalpic and kinetic measurements. Chem Listy (Prague). 1970;64:695–71 (in Czech).
Garn PD. Thermal analysis of investigation. New York: Academic Press; 1965.
Smothers WJ, Chiang Y. Handbook of DTA. New York: Chem. Publ; 1966.
Schultze D. Differential thermoanalyze. Berlin: VEB; 1969.
Mackenzie RC, editor. Differential thermal analysis, vol I and II. London: Academic; 1970 and 1972.
Smykats-Kloss W. Differential thermal analysis. Berlin: Springer; 1974.
Pope MI, Judd MD. Differential thermal analysis. London: Heyden; 1977.
Šesták J. Thermodynamic basis for the theoretical description and correct interpretation of thermoanalytical experiments. Thermochim Acta. 1979;28:197–227.
Šesták J. Differential thermal analysis, Chap. 12. In: Thermophysical Properties of Solids: theoretical thermal analysis. Amsterdam: Elsevier; 1984 and Russian translation Mir, Moscow 1988.
Eyraud MC. Appareil d’analyse enthalpique différentielle. C R Acad Sci. 1954;238:1511–2.
Watson ES, O’Neill MJ, Justin J, Brenner N. A DSC for quantitative differential thermal analysis. Anal Chem. 1964;36:1233–9.
Šesták J. Use of phenomenological enthalpy versus temperature diagram (and its derivative-DTA) for a better understanding of transition phenomena in glasses. Thermochim Acta. 1996;280/281:175–90.
Kozmidis-Petrovic A, Šesták J. Forty years of the Hruby’ glass-forming coefficient via DTA when comparing other criteria in relation to the glass stability and vitrification ability. J Therm Anal Calorim. 2012;110:997–1004 (with 80 useful references).
Šesták J, Kozmidis-Petrovic A, Živković Ž. Crystallization kinetics accountability and the correspondingly developed glass-forming criteria. J Miner Metall B. 2011;47(2):229–39.
Šesták J. Citation records and some forgotten anniversaries in thermal analysis. J Therm Anal Calorim. 2012;109:1–5 (with 85 useful references).
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Sánchez-Jiménez PE, Criado JM, Pérez-Maqueda LA. Kissinger kinetic analysis of data obtained under different heating schedule. J Therm Anal Calorim. 2008;94:427–32.
Piloyan GO, Ryabchikov IO, Novikova SO. Determination of activation energies of chemical reactions by DTA. Nature. 1966;3067:1229.
Gray AP. A simple generalized theory for analysis of dynamic thermal measurements. In: Porter RS, Johnson JF, editors. Analytical calorimetry, vol. 1. New York: Plenum Press; 1968. p. 209–18.
Gray AP. A generalized theory for analysis of dynamic thermal measurements. In: Buzas I, editor. Thermal analysis, Proceedings of the 4th ICTA. Budapest: Akademiai Kiado; 1974.
Holba P, Nevřiva M, Šesták J. Utilization of DTA for the determination of heats of the transformation of solid-solutions Mn3O4–Mn2CrO4. Izv. AN SSSR, Neorg. Materialy. 1974;10:2007–8 (in Russian).
Holba P, Šesták J, Bárta R. Theory and practice of DTA/DSC. Silikáty (Prague). 1976;20:83–95 (in Czech).
Šesták J, Holba P, Lombardi G. Quantitative evaluation of thermal effects: theory and practice. Ann Chim (Roma). 1977;67:73–87.
Nevřiva M, Holba P, Šesták J. Utilization of DTA for the determination of transformation heats. Silikáty (Prague). 1976;29:33–9 (in Czech).
Nevřiva M, Holba P, Šesták J. On correct measurements by means of DTA. In: Buzas I, editor. Thermal analysis, proceedings of 4th ICTA in Budapest. Budapest: Akademia Kiado; 1974. p. 981–90.
Šesták J, Holba P, Nevřiva M. Thermal inertia accounted in DTA evaluation. In: Dollimore D, editor. Thermal analysis, proceedings of the 2nd ESTAC. Salford: McMillan; 1976. p. 33–7.
Holba P, Nevřiva M. Description of thermoanalytical curves and the analysis of DTA peak by means of computer technique. Silikáty (Prague). 1977;21:19–23 (in Czech).
Holba P, Nevřiva M, Šesták J. Analysis of DTA curve and related calculation of kinetic data using computer technique. Thermochim Acta. 1978;23:223–31.
Höhne GWH, Hemminger W, Flammersheim HJ. Differential scanning calorimetry. Dordrecht: Springer; 2003. (readdition 2010).
Heines PJ, Reading M, Wilburn FW. Differential thermal analysis and differential scanning calorimetry. In: Brown ME, Gallagher PK, editors. Handbook of thermal analysis and calorimetry, vol. 1. Amsterdam: Elsevier; 2008. p. 279–361.
Boerio-Goates J, Callen JE. Differential thermal methods. In: Rossiter BW, Beatzold RC, editors. Determination of thermodynamic properties. New York: Wiley; 1992. p. 621–718.
Illekova E, Aba B, Kuhnast FA. Measurements of accurate specific heats of metallic glasses by DSC: analysis of theoretical principles and accuracies of suggested measurement procedure. Thermochim Acta. 1992;195:195–209.
Šesták J. Heat, thermal analysis and society. Hradec Kralove: Nucleus; 2004.
Šesták J. Science of Heat and Thermophysical Studies: a generalized approach to thermal analysis. Amsterdam: Elsevier; 2005.
Kubičár L, Illeková E. Use of pulse method for study of structural changes of materials. Thermochim Acta. 1985;92:441–44.
Kubičár L. Pulse methods of measuring basic thermophysical parameters. Amsterdam: Elsevier; 1990.
Holba P, Šesták J, Sedmidubský D. Heat transfer and phase transition at DTA experiments, Chap. 5. In: Šesták J, Šimon P, editors. Thermal analysis of micro-, nano- and non-crystalline materials. Berlin: Springer; 2013. p. 99–134. ISBN 978-90-481-3149-5.
Holba P, Šesták J, Sedmidubský D. DTA/DSC equation involving the effect of temperature gradient changes inside the sample. Chemické listy (Prague) 2012;106; 583-84 (in Czech).
Svoboda H, Šesták J. A new approach to DTA calibration by predetermined amount of Joule heat. In: Buzas I, editor. Thermal analysis, proceedings of the 4th ICTA. Budapest: Akademia Kiado; 1974. p. 726–31.
Svoboda H, Šesták J. Use of rectangular and triangular heat pulses in DTA analysis. In: TERMANAL, proceedings of the 9th TA conference at High Tatras. Bratislava: Publ. House SVŠT; 1973. pp. 12–7 (in Czech).
Carslaw HS, Jaeger JC. Conduction of heat in solids. Oxford: Oxford University Press; 1959. ISBN 978-0-19-853368-9.
Minakov AA, Adamovsky SA, Schick C. Non-adiabatic thin-film-chip nanocalorimetry. Thermochim Acta. 2005;432:177–85.
Adamovsky SA, Minakov AA, Schick C. Ultra-fast isothermal calorimetry using thin film sensors. Thermochim Acta. 2004;415:1–7.
Höhne GWH. On de-smearing of heat-flow curves in calorimetry. Thermochim Acta. 1978; 22:347–62.
Wang XY, Hom IK, Wu CN. Method for correcting the time-log of a conduction calorimeter and its application. Thermochim Acta. 1988;123:177–82.
Wiesner S, Woldt E. An algorithm for the reconstruction of the true specimen signal of a DSC calorimeter. Thermochim Acta. 1991;187:357–62.
Sarge SM, Gmelin E, Höhne GWH, Cammenga HK, Hemminger W, Eysel W. The caloric calibration of scanning calorimeters. Thermochim Acta. 1994;247:129–68.
Lölich KR. On the characteristics of the signal curves of heat flux calorimeters in studies of reaction kinetics. Part 1. A contribution to desmearing techniques. Thermochim Acta. 1994;231:7–20.
Kempen ATW, Sommer F, Mittemeijer EJ. Calibration and desmearing of DTA measurements signal upon heating and cooling. Thermochim Acta. 2002;383:21–30.
Tydlitát V, Zákoutský J, Černý R. Heat inertia correction of data measured by a conduction calorimeter. In: Thermophysics 2012, conference proceedings by Brno University of Technology, Brno 2012, pp. 235–40. ISBN: 978‐80‐214‐4599‐4.
Vonk CG. A procedure for desmearing X-ray scattering curves. J Appl Cryst. 1971;4:340–2.
Lifshin E. X-ray characterization of materials. New York: Wiley; 1999.
Lyon RE, Safronova N, Senese J, Stoliarov SI. Thermokinetic model of sample response in nonisothermal analysis. Thermochim Acta. 2012;545:82–9.
Šesták J, Holba P, Kratochvil J. Kinetics of thermal heterogeneous processes with the participation of solids. In: Pavlyuchenko MM, Prodan I, editors. Heterogeneous chemical reactions and reaction capability. Minsk: Nauka i Technika; 1975. p. 519–31 (in Russian).
Swarin SJ, Wims AM. Method for determining reaction kinetics by DSC. Anal Calorim. 1976;4:155–77.
Málek J. The kinetic analysis of non-isothermal data. Thermochim Acta. 1992;200:257–69.
Koga N. Physico-geometric kinetics of solid-state reactions by thermal analysis. J Therm Anal. 1997;49:45–56.
Vyazovkin S, Wight CA. Kinetic concepts of thermally stimulated reactions in solids: a view from a historical perspective. Int Rev Phys Chem. 2000;19:45–60.
Šimon P. Isoconversional kinetic methods: fundamentals, meaning and application. J Therm Anal Calorim. 2004;76:123–32.
Liu F, Sommer F, Bos C, Mittemeijer EJ. Analysis of solid state phase transformation kinetics: models and recipes. Int Mater Rev. 2007;52:193–9.
Moukina E. Determination of kinetic mechanism for reactions measured with thermal analysis. J Therm Anal Calorim. 2012;109:1203–14.
Šesták J. Rationale and fallacy of thermoanalytical kinetic patterns: how we model subject matter. J Therm Anal Calorim. 2012;110:5–16.
Šesták J. Philosophy of non-isothermal kinetics. J Therm Anal. 1979;16:503–20.
Šesták J, Holba P, Nevřiva M, Bergstein A. Kinetics of precipitation processes in the system Mn x Fe3−x O4. In: TERMANAL, proceedings of the 9th TA conference at High Tatras. Bratislava: SVST; 1973. p. S67–71 (in Czech).
Illeková E. On the various activation energies at crystallization of amorphous metallic materials. J Non-Cryst Solids. 1984;68:153–62.
Galwey AK. What theoretical and/or chemical significance is to be attached to the magnitude of an activation energy determined for a solid-state decomposition? J Therm Anal Calorim. 2006;86:267–86.
Brown ME, Maciejewski M, Vyazovkin S, Nomen R, Sempere J, Burnham A, Opfermann J, Strey R, Anderson HL, Kemmler A, Keuleers R, Janssens J, Desseyn HO, Li C-R, Tang TB, Roduit B, Malek J, Mitsuhashi T. Computational aspects of kinetic analysis Part A: the ICTAC kinetics project-data, methods and results. Thermochim Acta. 2000;355:125–43.
Vyazovkin S, Burnham AK, Criado JM, Pérez-Maqueda LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Škvára F, Šesták J. Computer calculation of the mechanism and associated kinetic data using a non-isothermal integral method. J Therm Anal Calorim. 1975;8:477–89.
Málek J. A computer program for kinetic analysis of non-isothermal thermoanalytical data. Thermochim Acta. 1989;138:337–46.
Chen D, Gao X, Dollimore D. Computer programs for kinetic analysis of non-isothermal TG data. Instrum Sci Technol. 1992;20:137–52.
Reading M, Elliot D, Hill VL. A new approach to the calorimetric investigation of physical and chemical transitives. J Therm Anal. 1993;40:949–55.
Wunderlich B, Jin Y, Boller A. Mathematical description of differential scanning calorimetry based on periodic temperature modulation. Thermochim Acta. 1994;238:277–93.
Málek J, Šesták J, Rouquerol F, Rouquerol J, Criado JM, Ortega A. Possibilities of two non-isothermal procedures (temperature- and/or rate-controlled) for kinetic studies. J Therm Anal. 1992;38:71–87.
Reading M, Price DM, Grandy D, Smith RM, Conroy M, Pollock HM. Microthermal analysis of polymers: current capabilities and future prospects. Macromol Symp. 2001;167:45–55.
Hammiche A, Reading M, Pollock HM, Song M, Hourston DJ. Localized thermal analysis using a miniaturized resistive probe. Rev Sci Instrum. 1996;67:4268–75.
Price DM, Reading M, Hammiche A, Pollock HM. Micro-thermal analysis: scanning thermal microscopy and localized thermal analysis. Int J Pharm. 1999;192:85–96.
Wunderlich B. Calorimetry of nanophases of macromolecules. Int J Thermophys. 2007;28:958–67.
Höhne GWH. Calorimetry on small systems—a thermodynamic contribution. Thermochim Acta. 2003;403:25–36.
Mareš JJ, Šesták J. An attempt at quantum thermal physics. J Therm Anal Calorim. 2005;82:681–6.
Volz S, editor. Microscale and nanoscale heat transfer. Heidelberg: Springer; 2007.
Nanda KK. Size-dependent melting of nanoparticles: hundred years of thermodynamic models. Pramana J Phys (India). 2009;72:517–628.
Guisbiers G, Buchaillot L. Universal size/shape-dependent law for characteristic temperatures. Phys Lett A. 2009;374:305–8.
Mayoral A, Barron H, Salas RE, Duran AV, Yacamán MJ. Nanoparticle stability from the nano- to the meso-interval. Nanoscale. 2010;2:335–42.
Levitas VI, Samani K. Size and mechanic effects in surface-induced melting of nanoparticles. Nat Commun. 2011;2.
Leitner J. Temperature of nanoparticles melting. Chem Listy (Prague). 2011;105:174–85 (in Czech).
Acknowledgments
The results were developed within the CENTEM project, reg. no. CZ.1.05/2.1.00/03.0088 that is co-funded from the ERDF within the OP RDI Program of the Ministry of Education, Youth and Sports Critical comments by David Sedmidubský, Emilia Illeková, Ludovít Kubičár, Jiří J. Mareš and Peter Šimon are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Šesták, J., Holba, P. Heat inertia and temperature gradient in the treatment of DTA peaks. J Therm Anal Calorim 113, 1633–1643 (2013). https://doi.org/10.1007/s10973-013-3025-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3025-3