
Abstract
Practical usefulness of the kinetic deconvolution for partially overlapped thermal decomposition processes of solids was examined by applying to the co-precipitated basic zinc carbonate and zinc carbonate. Comparing with the experimental deconvolutions by thermoanalytical techniques and mathematical deconvolutions using different statistical fitting functions, performance of the kinetic deconvolution based on an accumulative kinetic equation for the independent processes overlapped partially was evaluated in views of the peak deconvolution and kinetic evaluation. Two-independent kinetic processes of thermal decompositions of basic zinc carbonate and zinc carbonate were successfully deconvoluted by means of the thermoanalytical measurements in flowing CO2 and by applying sample controlled thermal analysis (SCTA). The deconvolutions by the mathematical curve fittings using different fitting functions and subsequent formal kinetic analysis provide acceptable values of the mass-loss fractions and apparent activation energies of the respective reaction processes, but the estimated kinetic model function changes depending on the fitting functions employed for the peak deconvolution. The mass-loss fractions and apparent kinetic parameters of the respective reaction processes can be optimized simultaneously by the kinetic deconvolution based on the kinetic equation through nonlinear least square analysis, where all the parameters indicated acceptable correspondences to those estimated through the experimental and mathematical deconvolutions. As long as the reaction processes overlapped are independent kinetically, the simple and rapid procedure of kinetic deconvolution is useful as a tool for characterizing the partially overlapped kinetic processes of the thermal decomposition of solids.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thermal decomposition of co-precipitated precursor solids is one of the common routes of ceramics processing. Experimental techniques of thermal analysis are the powerful tool for characterizing the mixed ratio of the component solids and the kinetic behaviors of the respective thermal decomposition steps. Because both the mixed ratio and kinetic behaviors influence largely on the characteristics of the product solids, monitoring of the thermoanalytical curves of the respective batches of the precursor solids are required for controlling the reaction processes and the quality of the product solids.
When the thermal decomposition processes of the component solids are overlapped partially on the thermoanalytical curves, the deconvolution of the overlapped processes are required to evaluate the mixed ratio of the precursor solids and the kinetic behaviors of the respective thermal decomposition steps. Various experimental techniques of thermal analysis can be applied to resolve the overlapped processes [1]. Among others, sample controlled thermal analysis (SCTA) [2, 3] can be utilized for this purpose. Peak fitting of the themoanalytical curves by mathematical functions are also applied to deconvolute the overlapped reaction processes as in the cases of chromatography and spectroscopy. Various mathematical functions have been utilized for the better peak fittings and successful deconvolution of the overlapped reaction processes [4–11]. For the deconvolution of the complex reaction processes of the thermal degradation of polymer, biomass, industrial wastes, and so on, an alternative deconvolution method has been applied by several groups [12–16], which has been carried out by applying directly the kinetic equation of multi-step reaction processes and optimizing all the necessary parameters through non-linear least square analysis. Because the kinetic deconvolution method is applicable for the overlapped reaction processes as long as those processes are independent kinetically, it is highly expected that some of the partially overlapped thermal decomposition steps in the ceramics processing can be solved by the method. At the same time, however, the kinetic deconvolution of the thermoanalytical curve is an advanced kinetic calculation by single run method, where mutual dependences of the kinetic parameters and the wrong estimation of the apparent kinetic parameters have to be always considered [17, 18].
This study was undertaken to evaluate the applicability of the kinetic deconvolution method to the partially overlapped thermal decomposition steps in the ceramics processing. Co-precipitated zinc carbonates, i.e., a mixture of basic zinc carbonate and zinc carbonate, were selected as model sample, because the thermal decompositions of these compounds are overlapped partially on the thermoanalytical curves and the mixed ratio changes depending on the precipitation conditions. By comparing with the conventional deconvolution methods of thermal analysis and mathematical peak fitting, characteristics of the kinetic deconvolution method applied to partially overlapped thermal decomposition steps in the ceramic processing are revealed.
Experimental
Sample preparation
All the chemicals utilized for preparing precipitates of zinc carbonates were reagent grade purchased from Sigma-Aldrich Japan. Every 1.0 mol dm−3 of stock solutions of zinc salts as listed in Table 1 were prepared by dissolving those chemicals into deionized-distilled water. The pH values of the stock solutions of zinc salts were measured by a pH meter (Horiba F-22). Similarly, a stock solution of sodium carbonate (1.0 mol dm−3) was also prepared. Every 500 cm3 of one of the stock solutions of zinc salt and sodium carbonate were mixed in a beaker and stirred mechanically at 350 rpm for 24 h at room temperature using a rotary impeller stirrer. The precipitates were separated by vacuum filtration and washed repeatedly with deionized-distilled water. The precipitates separated were dried in an electric oven at 353 K for 24 h. The samples obtained from different stock solutions of zinc salts were termed A–D as listed in Table 1.
Characterization
The samples were characterized by powder X-ray diffractometry (XRD), thermogravimetry–differential thermal analysis (TG–DTA), TG/DTA–mass spectroscopy (MS), and measurement of specific surface area. The powder XRD patterns of the samples were collected using a diffractometer (RINT 2200 V, Rigaku Co.) with monochrome Cu-Kα radiation (40 kV, 20 mA). TG–DTA measurements were performed for the samples of 10.0 mg, weighed into a platinum cell (6 mmϕ and 2.5 mm in height), in flowing N2 (80 cm3 min−1) at a heating rate β of 5 K min−1. For characterizing the evolved gases during the course of thermal decomposition, TG/DTA–MS (Thermomass, Rigaku Co.) was measured using an instrument constructed by coupling a TG–DTA (TG8120, Rigaku Co.) with a quadrupole mass spectrometer (M-200Q, Anelva), where the samples of 5.0 mg weighed into a platinum cell (5 mmϕ and 2.5 mm in height) were heated at β = 10 K min−1 in flowing He (200 cm3 min−1). The specific surface area of the samples was measured by the single-point method of BET (Flow SorbII-2300, Micromeritics Co.) after pretreatment of the samples at 353 K for 1 h.
For the sample obtained from the stock solution of zinc chloride, i.e., Sample C, further measurements of thermal analyses were carried out for characterizing the thermal decomposition process. For higher resolution of partially overlapped thermal decomposition processes of zinc carbonates, TG–DTA measurements were carried out in flowing N2–CO2 (80 % CO2) under the conditions otherwise identical to the above TG–DTA measurements in flowing N2. For the same purpose, two different techniques of SCTA were employed, i.e., the controlled transformation rate TG (CRTG) [19] and the controlled rate evolved gas analysis coupled with TG (CREGA–TG) [20]. CRTG measurement was performed by using a hanging type TG (TGA-50, Shimadzu Co.) equipped with a self-constructed SCTA controller [21–24]. The sample of 10.0 mg weighed into a platinum cell (6 mmϕ and 2.5 mm in height) was heated at β = 2 K min−1 in flowing N2 (80 cm3 min−1), while during the thermal decomposition reactions the mass-loss rate was regulated to be the constant rate C = 10 μg min−1. An instrument of CREGA-TG was constructed by connecting an infrared CO2 meter (LX-720, IIJIMA) and a hygrometer (HT20, NTK) to a TG–DTA (TGD5000, ULVAC) for monitoring the concentrations of CO2 and H2O in the outflow gas from the TG–DTA instrument during heating the sample. The sample of 10.0 mg weighed into a platinum cell (5 mmϕ and 5 mm in height) was heated at β = 2 K min−1 in flowing dry N2 (200 cm3 min−1), while during the thermal decomposition reactions CO2 concentration in the outflow gas was regulated to be the constant value of 50 ppm. For the purpose of kinetic analysis, the above mentioned TG–DTA measurements in flowing N2 were also carried out at different β, 1 ≤ β ≤ 10 K min−1.
Results and discussion
Sample characterization
Powder XRD patterns of the samples are shown in Fig. 1. All the major peaks of Sample A correspond to either Zn5(CO3)2(OH)6 (JCPDS 19-1458) or Zn4CO3(OH)6·H2O (JCPDS 11-0287) [25]. By the quantitative analysis of the evolved gases during the thermal decomposition by means of TG–MS, it has been revealed that the composition of Sample A corresponds to mineral hydrozincite (HZ), Zn5(CO3)2(OH)6 [26]. With increasing pH value of the stock solution of zinc salts, see Table 1, a systematic growth of the XRD peaks attributed to zinc carbonate (ZC), ZnCO3 (JCPDS 8-0449), is observed. It was reported that in the present system formation of ZC takes place from the mother solution with pH > 7.2 [27]. Because XRD peaks of HZ are observed even for Sample D, Samples B–D are identified as the mixtures of HZ and ZC. The specific surface area, S BET, of the respective samples is listed in Table 1. A higher value of S BET is observed for Samples A and B, while from Sample B to D the value decreases systematically with increasing the pH value of the mother solution and the content of ZC.

Typical XRD patterns of Samples A–D
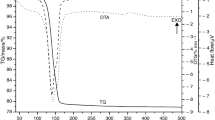
Typical TG–DTA curves of the samples are shown in Fig. 2. For Sample A, the TG–DTA traces indicate initial acceleration part and reaction tail, while the observed mass-loss, 26.4 ± 0.4 %, corresponds to the stoichiometric mass loss, 25.9 %, of the following reaction:

Typical TG–DTA curves for the thermal decomposition of Samples A–D (m 0 = 10.0 mg) at β = 5 K min−1 in flowing N2 (80 cm3 min−1)
It is clearly seen from the TG–DTA curves that the thermal decompositions of Samples B–D take place via two step reactions overlapped partially. The mass-loss due to the second reaction step increases from Sample B to D. It is reported that, in comparison with HZ, the thermal decomposition of ZC takes place at higher temperatures according to [28–31]:
It was also confirmed by powder XRD that the product solids of the thermal decomposition of Samples A–D are exclusively ZnO. Accordingly, the first and second reaction steps taking place during the thermal decomposition of the Samples B–D can be assigned to the reactions of Eqs. (1) and (2), respectively. This is more clearly seen in the mass chromatograms of m/z 18 and m/z 44 for the evolved gas during the thermal decompositions of Samples B–D shown in Fig. 3. Irrespective of the samples, the first reaction step is characterized by the simultaneous evolutions of CO2 and H2O as in Eq. (1). CO2 is the major gaseous product of the second reaction step, supporting Eq. (2). The detectable evolution of H2O at the second half stage of the second reaction step is likely due to the evolution of trapped water triggered by the crystal growth of ZnO. The change in the mass chromatogram of m/z 44 indicates the increase in the content of ZnCO3 from Sample B to D. From a view point of reaction kinetics, these two reaction steps can be recognized as the independent kinetic processes.

Mass chromatograms of m/z 18 and m/z 44 for the evolved gas during the thermal decompositions of Samples B–D (m 0 = 5.0 mg) at β = 10 K min−1 in flowing He (200 cm3 min−1)
Figure 4 shows TG–DTA curves for the thermal decomposition of Sample C at different β. Because the DTA peak tops of the respective reactions are separated enough, the apparent activation energy E a for the respective reaction steps is estimated roughly by the Kissinger method [32] from the relationship between the peak top temperature, T p, and β.
where α, α p, A, and f(α) are the fractional reaction, the fractional reaction at the peak top temperature, Arrhenius preexponetial factor, and kinetic model function, respectively. The Kissinger plots for the first and second reaction steps of the thermal decomposition of Sample C are shown in Fig. 5. From the slope of the plots, the values of E a, 143.2 ± 2.7 and 127.8 ± 7.3 kJ mol−1, were calculated for the first and second reaction steps, respectively.

TG–DTA curves for the thermal decomposition of Sample C (m 0 = 10.0 mg) at different β in flowing N2 (80 cm3 min−1)

Kissinger plots for the first and second step reactions of the thermal decomposition of Sample C
Experimental deconvolution
In Fig. 6, TG–DTG curves for the partially overlapped thermal decomposition processes of Sample C in flowing N2 and N2–CO2 (80 % CO2) are compared. By the effect of atmospheric CO2, TG–DTG curves shift to the higher temperatures as is expected from the chemical equilibrium in Eqs. (1) and (2). The respective reactions are separated each other in comparison with those in flowing N2. From the TG–DTG curves in flowing N2–CO2 (80 % CO2), the mass-loss fractions of the respective reactions of Eqs. (1) and (2) to the total mass-loss, c 1 and c 2, were estimated to be c 1 = 0.57 and c 2 = 0.43, respectively.

TG–DTG curves for the partially overlapped thermal decomposition processes of Sample C at β = 10 K min−1 in flowing N2 and N2–CO2 (80 % CO2)
Figure 7 shows typical results of SCTA for the thermal decomposition of Sample C under constant mass-loss rate (a) and under constant evolution rate of CO2 (b). In these SCTA records, the respective reactions of Eqs. (1) and (2) are separated clearly by the temperature jump on the way of the thermal decomposition. The values of c 1 and c 2 evaluated by SCTA measurements corresponded exactly to those determined from the TG–DTG curves in flowing N2–CO2 (80 % CO2).

Typical results of SCTA for the thermal decomposition of Sample C under constant mass-loss rate (a) and under constant evolution rate of CO2 (b)
The practically identical values of c 1 and c 2 evaluated by the above three different measurements indicate successful deconvolution of the overlapped processes by those experimental techniques. As can be seen for the change in the shape and position of the mass-loss curves by the effect of atmospheric CO2 in Fig. 6, however, the successful deconvolution is due to the change in the kinetic behavior of the respective reactions by an effect of experimental condition other than temperature. For the restricted purpose of the simultaneous characterizations of sample composition and kinetics of the respective reaction steps under the selected reaction condition, such changes of kinetic behaviors make it difficult to characterize the kinetic process under certain condition predetermined. In this sense, the experimental deconvolution is not always the successful method for the present purpose.
Mathematical deconvolution
After examining the applicability of various fitting functions empirically, nine mathematical functions, i.e., asymmetric double sigmoid (ADS), asymmetric logistic (AL), extreme value 4 parameter fronted (EV4PF), Fraser-Suzuki (FS) [33, 34], log normal 4 parameter (LN4P), logistic power peak (LPP), Pearson IV (P-IV), Pearson IV (a3 = 2) (P-IV(a3 = 2)), and Weibull (WB) were selected as the possible fitting functions with R 2 > 0.99 for the deconvolution of the DTG curves of the thermal decomposition of Sample C at different β. The mathematical forms of these fitting functions were listed in “Electronic Supporting Material”. The fitting procedure was performed using commercially available software, PeakFit ver4.12. Only the TG–DTG curves at β ≤ 5 K min−1 were subjected to the peak deconvolution and subsequent kinetic analysis, because a distinguishable change of the performance of the peak fitting was observed for the TG–DTG curves recorded at β > 5 K min−1, probably due to the influence of heat and mass-transfer phenomena that become relevant for linear heating rate experiments under higher heating rates. This assumption is supported by the influence of CO2 on the present thermal decomposition process observed in Fig. 6 and reported complex kinetic behavior of the thermal decomposition of HZ under high water vapor pressure [35] and under self-generated water vapor pressure [36].
Figure 8 compares typical results of peak fittings by using ADS and WB functions. Irrespective of fitting functions employed, the attributions R 2 estimated were better than 0.99 when deconvoluted into two reaction peaks. The fitting performance of the respective fitting functions can be categorized into three different types. Comparing the fitted curves by ADS and WB, the large difference can be found at the peak tails in both sides. The peak tails of ADS are apparently longer than those of WB. The functions which indicate the longer and the shorter peak tails were labeled as types I and II, respectively, together with the intermediate type III. The types of the respective fitting functions are summarized in Table 2. In addition, a distinguishable residue at the initial part of the first reaction step is observed irrespective of the fitting functions employed. This is apparently due to the surface reaction of the thermal decomposition of HZ [35, 36]. Addition of another fitting peak at the initial part of the first reaction peak improves slightly the fitting results, but this procedure deforms the fitting curves for the established reaction of the first reaction step. This is not favorable for the subsequent kinetic calculation of the deconvoluted curves.

Typical results of mathematical deconvolution of DTG curve for Sample C at β = 2 K min−1 by using ADS and WB functions
As an example, TG–DTG curves deconvoluted into two peaks by using the ADS function are shown in Fig. 9. The values of c 1 and c 2 evaluated from the TG curves deconvoluted using different fitting functions are listed in Table 2. The value of c 1 indicates slightly lower value than that determined by the experimental deconvolution above. When the DTG curves were fitted by assuming three-independent peaks, that is, two peaks for the first reaction step and one peak for the second reaction step, the correspondence of values of c 1 and c 2 to those determined experimentally was improved.

TG–DTG curves for the respective reaction steps of the thermal decomposition of Sample C at different β deconvoluted by using ADS function
The deconvoluted TG–DTG curves for the thermal decomposition of Sample C at different β were subjected to the formal kinetic analysis by assuming the following kinetic equation:
The apparent values of E a at different α were evaluated by the Friedman method [37] through plotting ln(dα/dt) against T −1. Figure 10 compares the values of E a evaluated from the respective TG–DTG curves deconvoluted using ADS and WB functions. It is apparent that the values of E a change depending on α, although the variation is within 10 % of the averaged value. The behavior of α-dependent change is different between the fitting functions employed. Nevertheless, the values of E a averaged over the main reaction part (0.1 ≤ α ≤ 0.9) are comparable among all the fitting functions as listed in Table 2. The values of E a evaluated for the respective reaction steps are slightly different from those evaluated by the Kissinger plots shown in Fig. 5. The differences are interpreted by taking account the approximation of the Kissinger methods for applying to the rate process other than that of the first-order law [38, 39], difference in the range of heating rate applied to the calculation and the possible influence of the partial overlapping of the two reaction steps on the T p of the respective reaction steps in Fig. 4.

Comparisons of the values of E a evaluated for the thermal decomposition of Sample C from the TG–DTG curves deconvoluted by using ADS and WB functions
Assuming the averaged E a values in the range 0.1 ≤ α ≤ 0.9, the experimental master plot of the reaction rate extrapolated to infinite temperature (dα/dθ) against α were drawn by calculating the values of (dα/dθ) at different α according to [40–42]:
where θ is the Ozawa’s generalized time denoting the reaction time extrapolated to infinite temperature [43, 44]. Figure 11 compares the experimental master plots drawn for the TG–DTG curves deconvoluted using the fitting functions of ADS and WB. Shapes of the experimental master plots are largely different between those drawn for the TG–DTG curves deconvoluted using different functions and classified according to the types of fitting functions categorized in Table 2. The experimental master plots were correlated to the kinetic model function f(α) according to [40–42]:

Comparisons of the experimental master plots of (dα/dθ) against α evaluated for the thermal decomposition of Sample C from the TG–DTG curves deconvoluted by using ADS and WB functions, together with the fitting curves by SB(m,n,p) model
An empirical kinetic model function known as Sestak–Berggren model [45]: SB(m,n,p), f(α) = α m(1-α)n[−ln(1−α)]p, was employed as the most appropriate one for the present purpose of the kinetic fitting, because of the high flexibility of the function to fit various types of physico-geometrical mechanisms of the solid-state reaction [46–49] and those deviated cases with nonintegral or fractal reaction geometry [50–52], size distribution of the reactant particles [53], and so on [54]. The high flexibility makes it difficult to interpret the physico-chemical meanings of the kinetic exponent, but SB model with the three parameters indicate the highest performance as a fitting function required in this study. Through the optimizations of the kinetic exponents in SB(m,n,p) and the preexponential factor A by non-linear regression analysis based on the Levenberg–Marquardt algorithm, the kinetic exponents of SB(m,n,p) and the preexponential factor A summarized in Table 2 were evaluated, giving the fitting curves drawn in Fig. 11. The values of kinetic exponents and A are largely different among the experimental master plots drawn from the TG–DTG curves deconvoluted using the respective fitting functions and are classified according to the types of fitting functions categorized as Types I–III.
The above results demonstrate that the mass-loss fractions of the respective reaction steps can be estimated properly by the curve fittings using any appropriate fitting functions. However, the trend of α-dependent change in E a, shape of the experimental master plot, and the kinetic exponents estimated change depending on the fitting function employed. For the purpose of kinetic analysis, the correspondence of the fitting function to the kinetic behaviors of the respective thermal decomposition steps should be considered carefully.
In spite of the α-dependent change of E a values evaluated by the Friedman method, the averaged values of E a in the main part of reaction are comparable among those values evaluated from the TG–DTG curves deconvoluted using the different fitting functions. This seems to be due to that the main part of the DTG peaks of the respective reaction steps are fitted nearly perfectly and the fitting performance does not change among the deconvoluted DTG curves at different β. Accordingly, application of the Kissinger plot to the deconvoluted DTG curves at different β is a good approximation for evaluating the apparent values of E a for the respective reaction steps overlapped partially. The values of E a evaluated by the Kissinger plots for the deconvoluted DTG curves of the respective reaction steps were in agreement with those evaluated by the Friedman plots within the standard error.
Kinetic deconvolution
A direct application of the kinetic equation to the peak separation may avoid the empirical selection of the fitting function. As long as the overall process of the thermal decomposition is composed of N independent kinetic processes, the overall kinetic behavior is expressed by the summation of the respective kinetic processes i by considering their mass-loss fractions, c i [13, 15, 16].
The empirical SB(m,n,p) is assumed as the kinetic model functions of the respective reaction steps, f i(α i). The most appropriate parameters, c i, A i, E a,i, m i, n i, and p i, of the respective reaction steps are optimized simultaneously by the nonlinear least square analysis to be minimizing the square sum of the residue when fitting the experimental curve of (dα/dt)exp versus time by the calculated curve of (dα/dt)cal versus time [13, 15, 16].
In this reaction system, totally 12 parameters are optimized when assuming the independent two step thermal decomposition reactions. In this type of parameter optimization by non-linear least square analysis, it is always required to set the appropriate initial values of all the parameters [55, 56], in order to avoid the apparent solutions by the local minimum of the F value. In this study, the initial values of c i, A i, and E a,i were selected by referencing the above results of experimental and mathematical deconvolutions. For the kinetic model function f i(α i), the first order reaction, that is, SB(0,1,0), was selected for both the reaction steps as the initial setting.
Figure 12 shows typical results of the kinetic deconvolution for the thermal decomposition of Sample C. As in the case of mathematical peak fitting, a distinguishable residue is observed at the initial part of the first decomposition step. The R 2 value averaged over those obtained for the thermal decomposition at different β was 0.984 ± 0.005. Table 3 summarizes the optimized parameters for the thermal decomposition of Sample C at different β. The values of c 1 and c 2 are comparable with those estimated by the mathematical peak fittings assuming two peaks and indicate the constant values at different β. The Arrhenius parameters, A and E a, optimized for the respective reaction steps are practically constant irrespective of β. The value of E a for the first reaction step is the intermediate value of those determined by the Kissinger method and from the mathematically deconvoluted DTG curves. The value for the second reaction step is nearly corresponding to that determined by the Kissinger method. Although the kinetic exponents in SB(m,n,p) deviate slightly among the values determined at different β, the deviations are acceptable by considering the high sensitivity of the empirical kinetic model with three kinetic exponents. The positive values of m are different from those evaluated by the formal kinetic analysis of the mathematically deconvoluted DTG curves in Table 2, but the differences are compensated by the smaller values of p. The profile of SB(m,n,p) model optimized by the kinetic deconvolution is nearly corresponding to those determined by the formal kinetic analysis of the DTG curves deconvoluted using the mathematical fitting functions of Type II. This is, in turn, supporting the reported successful mathematical deconvolutions and kinetic characterizations for the partially overlapped thermal decomposition processes of solids using the fitting functions of FS [11] and WB [6, 8, 10].

Typical result of the kinetic deconvolution for the thermal decomposition of Sample C at β = 5 K min−1
Using the kinetic parameters optimized by the kinetic deconvolution, the experimental master plots for the respective reaction steps are drawn in Fig. 13. The experimental master plots of (dα/dθ) against α for the respective reaction steps indicated the peak maximum on the way of the reaction, denoting the nucleation and growth type reactions as the possible physico-geometrical reaction model. The Johnson–Mehl–Avrami model of typical physico-geometrical model of nucleation and growth, JMA(m) [57]: f(α) = m(1−α)[−ln(1−α)]1−1/m, was applied to fit the experimental master plots according to Eq. (8). The experimental master plot of the first reaction step cannot be fitted by JMA(m), but the linear correlation between (dα/dθ) and α during the second-half of the reaction indicates the first-order rate behavior, that is, m = 1 in JMA(m). It is apparent that the solution of the problem on the initial reaction process of the first reaction step which could not be fitted satisfactorily both by the mathematical peak fitting and kinetic deconvolution is necessary condition for the rigorous kinetic description of the first reaction step, that is, the thermal decomposition of synthetic HZ. On the other hand, the experimental master plot of the second reaction step is fitted nearly perfectly by JMA(m) with m = 1.48 ± 0.01 (R 2 = 0.9931). The kinetic model JMA(1.5) is interpreted formally by the random nucleation and subsequent 1-D growth controlled by diffusion or by the instantaneous nucleation and 3-D growth controlled by diffusion. The detailed kinetic analyses for the thermal decompositions of synthetic HZ and ZC will be reported separately.

The experimental master plots of (dα/dθ) against α evaluated for the thermal decomposition of Sample C from the results of kinetic deconvolution, together with the fitting curves by JMA(m) model
Adopting the procedure of the kinetic deconvolution, the kinetic parameters of the thermal decomposition process of Samples A–D were evaluated, together with the values of c 1 and c 2. Table 4 summarizes the results of the kinetic deconvolution for the thermal decompositions of Samples A–D at β = 5 K min−1. The values of c 1 and c 2 change systematically from A to D, where the values optimized for the respective samples were in good agreement with those estimated by the mathematical peak deconvolution using WB function. In both the reaction steps, the kinetic parameters change systematically from A to C, but the comparable kinetic parameters were optimized for the Samples C and D. It is estimated from the results that from A to C both the mixed ratio of HZ and ZC phases and those particle characteristics which influence on the thermal decomposition kinetics are changing systematically, whereas from C to D only the mixed ratio is changing.
The similar situations of the partially overlapped thermal decomposition processes of the precursor solids can be found in elsewhere for the ceramics processing through thermal decomposition. It is very probable that the information evaluated through the rapid kinetic deconvolution of the partially overlapped thermal decomposition processes is useful as an analytical tool for characterizing the reactant solids and for controlling the characteristics of product solids. At the same time, however, the parameters optimized by the kinetic deconvolution based on Eq. (9) should be critically evaluated for maintaining the reliability as the apparent kinetic parameters. The experimental techniques of thermal analysis and the mathematical methods of peak fitting using appropriate statistical functions provide useful information for setting the initial parameters for the kinetic deconvolution and for evaluating the reliability of the optimized parameters.
Conclusions
In addition to the experimental techniques of thermal analysis and the mathematical methods of peak fitting for the deconvolution of partially overlapped thermal decomposition processes of solids, the direct application of the kinetic equation and optimization of the composition of the reactant mixture and the kinetic parameters of the respective reaction steps, i.e., kinetic deconvolution, is the other promising method when the overlapped reaction processes are unambiguously the independent kinetic processes. In view of the kinetic characterizations for the respective reactions under the fixed reaction conditions, the kinetic deconvolution is the simple and rapid procedure with some merits over the conventional experimental techniques and mathematical peak fitting. From the results of the kinetic deconvolution, the composition of the reactant mixture and the kinetic parameters of the respective thermal decomposition processes are evaluated simultaneously. As was demonstrated for the partially overlapped thermal decomposition processes of the co-precipitated zinc carbonates, the results optimized by the kinetic deconvolution are nearly in agreement with those evaluated by the experimental techniques and mathematical methods of deconvolution. By comparing the results of kinetic deconvolution among the thermal decomposition process of different batches of the precursor solids, possible changes in the mixed ratio of the precursor solids and those particle characteristics which influence on the thermal decomposition kinetics can be detected simultaneously.
References
Sorai M, editor. Comprehensive handbook of calorimetry & thermal analysis. Chichester: Wiley; 2004.
Sørensen OT, Rouquerol J, editors. Sample controlled thermal analysis. Dordrecht: Kluwer; 2003.
Sanchez-Jimenez PE, Perez-Maqueda LA, Crespo-Amoros JE, Lopez J, Perejon A, Criado JM. Quantitative characterization of multicomponent polymers by sample-controlled thermal analysis. Anal Chem. 2010;82(21):8875–80. doi:10.1021/ac101651g.
Bernard S, Fiaty K, Cornu D, Miele P, Laurent P. Kinetic modeling of the polymer-derived ceramics route: investigation of the thermal decomposition kinetics of poly[B-(methylamino)borazine] precursors into boron nitride. J Phys Chem B. 2006;110(18):9048–60. doi:10.1021/jp055981m.
Ozao R, Nishimoto Y, Pan W, Okabe T. Thermoanalytical characterization of carbon/carbon hybrid material, apple woodceramics. Thermochim Acta. 2006;440(1):75–80. doi:10.1016/j.tca.2005.10.014.
Cai J, Liu R. Weibull mixture model for modeling nonisothermal kinetics of thermally stimulated solid-state reactions: application to simulated and real kinetic conversion data. J Phys Chem B. 2007;111(36):10681–6. doi:10.1021/jp0737092.
Koga N, Yamane Y. Effect of mechanical grinding on the reaction pathway and kinetics of the thermal decomposition of hydromagnesite. J Therm Anal Calorim. 2008;93(3):963–71. doi:10.1007/s10973-007-8616-4.
Cai J, Alimujiang S. Kinetic analysis of wheat straw oxidative pyrolysis using thermogravimetric analysis: statistical description and isoconversional kinetic analysis. Ind Eng Chem Res. 2009;48(2):619–24. doi:10.1021/ie801299z.
Vecchio S, Cerretani L, Bendini A, Chiavaro E. Thermal decomposition study of monovarietal extra virgin olive oil by simultaneous thermogravimetry/differential scanning calorimetry: relation with chemical composition. J Agric Food Chem. 2009;57(11):4793–800. doi:10.1021/jf900120v.
Janković B, Adnađević B, Kolar-Anić L, Smičiklas I. The non-isothermal thermogravimetric tests of animal bones combustion. Part II. Statistical analysis: application of the Weibull mixture model. Thermochim Acta. 2010;505(1–2):98–105. doi:10.1016/j.tca.2010.04.005.
Perejon A, Sanchez-Jimenez PE, Criado JM, Perez-Maqueda LA. Kinetic analysis of complex solid-state reactions. A new deconvolution procedure. J Phys Chem B. 2011;115(8):1780–91. doi:10.1021/jp110895z.
Mamleev V, Bourbigot S, Le Bras M, Duquesne S, Šesták J. Thermogravimetric analysis of multistage decomposition of materials. Phys Chem Chem Phys. 2000;2(20):4796–803. doi:10.1039/b004357p.
Ferriol M, Gentilhomme A, Cochez M, Oget N, Mieloszynski JL. Thermal degradation of poly(methyl methacrylate) (PMMA): modelling of DTG and TG curves. Polym Degrad Stab. 2003;79(2):271–81. doi:10.1016/S0141-3910(02)00291-4.
Font R, Conesa JA, Moltó J, Muñoz M. Kinetics of pyrolysis and combustion of pine needles and cones. J Anal Appl Pyrol. 2009;85(1–2):276–86. doi:10.1016/j.jaap.2008.11.015.
Lopez G, Aguado R, Olazar M, Arabiourrutia M, Bilbao J. Kinetics of scrap tyre pyrolysis under vacuum conditions. Waste Manag. 2009;29(10):2649–55. doi:10.1016/j.wasman.2009.06.005.
Sánchez-Jiménez PE, Perejón A, Criado JM, Diánez MJ, Pérez-Maqueda LA. Kinetic model for thermal dehydrochlorination of poly(vinyl chloride). Polymer. 2010;51(17):3998–4007. doi:10.1016/j.polymer.2010.06.020.
Koga N, Sestak J, Malek J. Distortion of the Arrhenius parameters by the inappropriate kinetic-model function. Thermochim Acta. 1991;188(2):333–6. doi:10.1016/0040-6031(91)87091-a.
Koga N. A review of the mutual dependence of Arrhenius parameters evaluated by the thermoanalytical study of solid-state reactions: the kinetic compensation effect. Thermochim Acta. 1994;244(1):1–20. doi:10.1016/0040-6031(94)80202-5.
Koga N, Criado JM, Tanaka H. Kinetic analysis of the thermal decomposition of synthetic malachite by CRTA. J Therm Anal Calorim. 2000;60(3):943–54. doi:10.1023/a:1010172111319.
Koga N, Yamada S. Influences of product gases on the kinetics of thermal decomposition of synthetic malachite evaluated by controlled rate evolved gas analysis coupled with thermogravimetry. Int J Chem Kinet. 2005;37(6):346–54. doi:10.1002/kin.20089.
Alcalá M, Criado JM, Gotor FJ, Ortega A, Perez Maqueda LA, Real C. Development of a new thermogravimetric system for performing constant rate thermal analysis (CRTA) under controlled atmosphere at pressures ranging from vacuum to 1 bar. Thermochim Acta. 1994;240(1):167–73. doi:10.1016/0040-6031(94)87038-1.
Koga N, Criado JM. The influence of mass transfer phenomena on the kinetic analysis for the thermal decomposition of calcium carbonate by constant rate thermal analysis (CRTA) under vacuum. Int J Chem Kinet. 1998;30(10):737–44. doi:10.1002/(sici)1097-4601(1998)30:10<737:aid-kin6>3.0.co;2-w.
Perez-Maqueda LA, Criado JM, Gotor FJ. Controlled rate thermal analysis commanded by mass spectrometry for studying the kinetics of thermal decomposition of very stable solids. Int J Chem Kinet. 2002;34(3):184–92. doi:10.1002/kin.10042.
Criado JM, Pérez-Maqueda LA, Diánez MJ, Sánchez-Jiménez PE. Development of a universal constant rate thermal analysis system for being used with any thermoanalytical instrument. J Therm Anal Calorim. 2007;87(1):297–300. doi:10.1007/s10973-006-7813-x.
Kanari N, Mishra D, Gaballah I, Dupre B. Thermal decomposition of zinc carbonate hydroxide. Thermochim Acta. 2004;410(1–2):93–100. doi:10.1016/S0040-6031(03)00396-4.
Koga N, Tanaka H. Thermal decomposition of Copper(II) and zinc carbonate hydroxides by means of TG-MS—quantitative analyses of evolved gases. J Therm Anal and Calorim. 2005;82(3):725–9. doi:10.1007/s10973-005-0956-3.
Hales MC, Frost RL. Synthesis and vibrational spectroscopic characterisation of synthetic hydrozincite and smithsonite. Polyhedron. 2007;26(17):4955–62. doi:10.1016/j.poly.2007.07.002.
Gotor FJ, Macías M, Ortega A, Criado JM. Simultaneous use of isothermal, nonisothermal, and constant rate thermal analysis (CRTA) for discerning the kinetics of the thermal dissociation of smithsonite. Int J Chem Kinet. 1998;30(9):647–55. doi:10.1002/(SICI)1097-4601(1998)30:9<647:AID-KIN6>3.0.CO;2-S.
Budrugeac P, Criado JM, Gotor FJ, Popescu C, Segal E. Kinetic analysis of dissociation of smithsonite from a set of non-isothermal data obtained at different heating rates. J Therm Anal Calorim. 2001;63(3):777–86. doi:10.1023/A:1010148306206.
Vágvölgyi V, Hales M, Martens W, Kristóf J, Horváth E, Frost RL. Dynamic and controlled rate thermal analysis of hydrozincite and smithsonite. J Therm Anal Calorim. 2008;92(3):911–6.
Hales MC, Frost RL. Thermal analysis of smithsonite and hydrozincite. J Therm Anal Calorim. 2008;91(3):855–60. doi:10.1007/s10973-007-8571-0.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29(11):1702–6.
Fraser RDB, Suzuki E. Resolution of overlapping absorption bands by least squares procedures. Anal Chem. 1966;38(12):1770–3. doi:10.1021/ac60244a038.
Fraser RDB, Suzuki E. Resolution of overlapping bands. Functions for simulating band shapes. Anal Chem. 1969;41(1):37–9. doi:10.1021/ac60270a007.
Koga N, Tatsuoka T, Tanaka Y, Yamada S. Catalytic action of atmospheric water vapor on the thermal decomposition of synthetic hydrozincite. Trans Mater Res Soc Jpn. 2009;34(2):343–6.
Yamada S, Tsukumo E, Koga N. Influences of evolved gases on the thermal decomposition of zinc carbonate hydroxide evaluated by controlled rate evolved gas analysis coupled With TG. J Therm Anal Calorim. 2009;95(2):489–93. doi:10.1007/s10973-008-9272-z.
Friedman HL. Kinetics of thermal degradation of cha-forming plastics from thermogravimetry, application to a phenolic plastic. J Polym Sci C. 1964;6:183–95.
Criado J, Ortega A. Non-isothermal transformation kinetics: remarks on the Kissinger method. J Non-Cryst Solids. 1986;87(3):302–11. doi:10.1016/s0022-3093(86)80004-7.
Budrugeac P, Segal E. Applicability of the Kissinger equation in thermal analysis. J Therm Anal Calorim. 2007;88(3):703–7. doi:10.1007/s10973-006-8087-z.
Ozawa T. Applicability of Friedman plot. J Therm Anal. 1986;31:547–51.
Koga N. Kinetic-analysis of thermoanalytical data by extrapolating to infinite temperature. Thermochim Acta. 1995;258:145–59. doi:10.1016/0040-6031(95)02249-2.
Gotor FJ, Criado JM, Malek J, Koga N. Kinetic analysis of solid-state reactions: the universality of master plots for analyzing isothermal and nonisothermal experiments. J Phys Chem A. 2000;104(46):10777–82. doi:10.1021/jp0022205.
Ozawa T. A new method of analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38(11):1881–6. doi:10.1246/bcsj.38.1881.
Ozawa T. Non-isothermal kinetics and generalized time. Thermochim Acta. 1986;100(1):109–18. doi:10.1016/0040-6031(86)87053-8.
Sestak J, Berggren G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim Acta. 1971;3:1–12. doi:10.1016/0040-6031(71)85051-7.
Šesták J. Diagnostic limits of phenomenological kinetic models introducing the accommodation function. J Therm Anal. 1990;36(6):1997–2007. doi:10.1007/bf01914116.
Málek J. The kinetic analysis of non-isothermal data. Thermochim Acta. 1992;200:257–69. doi:10.1016/0040-6031(92)85118-f.
Perez-Maqueda LA, Criado JM, Sanchez-Jimenez PE. Combined kinetic analysis of solid-state reactions: a powerful tool for the simultaneous determination of kinetic parameters and the kinetic model without previous assumptions on the reaction mechanism. J Phys Chem A. 2006;110(45):12456–62. doi:10.1021/jp064792g.
Šimon P. Fourty years of the Šesták–Berggren equation. Thermochim Acta. 2011;520(1–2):156–7. doi:10.1016/j.tca.2011.03.030.
Ozao R, Ochiai M. Fractal reaction in solids. J Ceram Soc Jpn. 1993;101(11):263–7. doi:10.2109/jcersj.101.263.
Koga N. Accommodation of the actual solid-state process in the kinetic-model function. 1. Significance of the nonintegral kinetic exponents. J Therm Anal. 1994;41(2–3):455–69. doi:10.1007/bf02549327.
Koga N, Malek J. Accommodation of the actual solid-state process in the kinetic model function. 2. Applicability of the empirical kinetic model function to diffusion-controlled reactions. Thermochim Acta. 1996;283:69–80.
Koga N, Criado JM. Kinetic analyses of solid-state reactions with a particle-size distribution. J Amer Ceram Soc. 1998;81(11):2901–9. doi:10.1111/j.1151-2916.
Koga N, Tanaka H. A physico-geometric approach to the kinetics of solid-state reactions as exemplified by the thermal dehydration and decomposition of inorganic solids. Thermochim Acta. 2002;388(1–2):41–61. doi:10.1016/s0040-6031(02)00051-5.
de Levie R. Collinearity in least-squares analysis. J Chem Educ. 2012;89(1):68–78. doi:10.1021/ed100947d.
de Levie R. Nonisothermal analysis of solution kinetics by spreadsheet simulation. J Chem Educ. 2012;89(1):79–86. doi:10.1021/ed100948n.
Khawam A, Flanagan DR. Solid-state kinetic models: basics and mathematical fundamentals. J Phys Chem B. 2006;110(35):17315–28. doi:10.1021/jp062746a.
Acknowledgements
This study was supported partially by the grant-in-aid for scientific research (B) (21360340 and 22300272) from Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Koga, N., Goshi, Y., Yamada, S. et al. Kinetic approach to partially overlapped thermal decomposition processes. J Therm Anal Calorim 111, 1463–1474 (2013). https://doi.org/10.1007/s10973-012-2500-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2500-6