
Abstract
Nitro compounds are capable of rapid chemical decompositions with a large amount of energy releases and hence pose significant thermal explosion hazards. Molecular simulation has been well established and demonstrated as an effective tool to predict physical and/or chemical properties of energetic materials, such as onset temperature, heat of reaction, and shock sensitivity. In this work, a simple relationship for predicting the onset temperature of nitro aromatic compounds containing other functional groups is developed based on their molecular structures. The results have shown that the thermal onset temperature of a specific nitro aromatic compound is strongly related to its excitation energy (a singlet state to triplet state). The predicted onset temperatures show very good agreement with respect to the measured onset temperatures by differential scanning calorimetry. Deviations compared to the experimental values are very small. These correlations can be used to computationally screen new nitro compounds for their thermal explosion hazards. These correlations can also be applied as a preliminary thermal analysis method and expedite the evaluation process of new energetic materials.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thermal hazards are of great importance to the safer operation of chemical processes and now still remain the most serious concern in the chemical industry in spite of continued efforts that have devoted to this area in the last decades [1–3]. The United State Chemical Safety and Hazard Investigation Board (US CSB) reported 167 reactive chemical incidents between 1980 and 2001 [4]. In this report, energetic materials (or nitro compounds) were considered as typical reactive chemicals. Energetic materials, which contain multiple NO2 functional groups, are always used as fuels, high explosives, and propellants [5, 6]. Those nitro compounds are capable of rapid chemical decomposition with large energy release, triggering deflagrations and/or detonations and causing sever damages to people, property, and environment. Therefore, to prevent unexpected deflagrations and/or detonations, it is essential to investigate thermal hazards of nitro compounds, especially their initial decomposition temperature (i.e., onset temperature) and shock sensitivity [7].
The thermal hazard of a compound is an inherent property of the compound while the characterization of the thermal hazard is considered as a dynamic problem [8]. Usually both thermodynamic and kinetic data are necessary to be collected to evaluate the thermal hazards. Ando et al. [9] reported that onset temperature and heat of reaction are two significant parameters of thermal hazards, which correspond to their kinetic and thermodynamic properties, respectively. It is well known that onset temperatures are very difficult to be obtained or predicted. The onset temperature may be estimated by some of the experiments (i.e., differential scanning calorimetry (DSC) measurements) with a good calibration, but not with a great accuracy as would be desirable [10]. In addition, estimations of onset temperatures based on calorimetric experiments can be resource consuming. Therefore, it is necessary to find a way to calculate onset temperatures, especially for those highly reactive compounds that experiments are difficult to carry out [11].
For nitro compounds, in general, the initial steps in the thermal reactions are considered very important in the explosion, which can be caused by a thermal heat, shock, or mechanical impact [12]. Once the initial steps are triggered, the compound undergoes a rapid chemical reaction. The initial reactions have been studied for several typical energetic materials, such as RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine) [13], FOX-7 (1,1-diamino-2,2-dinitroethylene) [14], and TNAZ (1,3,3-trinitroazetidine) [15]. The rupture of the weakest C–NO2 bond as depicted in Eq. (1) is often suggested as an initial step in the thermal explosion of nitro compounds [16, 17]. Saraf et al. [18] have suggested that the onset temperatures of nitro compounds are closely related to their initial decomposition steps, especially the C–NO2 bond dissociation energy (BDE)
Our previous research has shown that the rupture of C–NO2 bond in nitroethane is a two-step process: (1) singlet to triplet excitation; (2) further excitation to a transition state [19]. Manaa et al. [20] also reported similar results in nitromethane by the density functional theory (DFT) calculations. For this two-step process, the first step is critical which usually requires more energy to excite electrons from a singlet state to triplet state (i.e., excitation energy). To better understand the relationship between excitation energy and onset temperature for nitro compounds, molecular simulation studies have been conducted on a certain group of nitro aromatic compounds in this work. The purpose of this work is to introduce simple correlations for the prediction of onset temperature of nitro aromatics. The predicted results are compared with experimental values as well as the previously developed correlation. It is shown here how this simple method can quickly predict reliable results on thermal onset temperature and screen notional energetic materials.
Data collection
The accuracy of a correlation is directly affected by the quality of the experimental data. It is well known that onset temperatures are experimentally determined data and the values reported by different authors as well as different organizations may differ a lot. For this reason, in order to have homogenous experimental data, all the onset temperature data of nitro compounds were collected from two previously published papers respectively. The first data set of 12 nitro compounds as shown in Table 1 was taken from the paper published by Duh et al. [21]. Duh’s data were obtained from the dynamic temperature scanning (4 °C min−1) on a Mettler TA4000 system coupled with a DSC25 measuring cell. The second data set of 18 nitro compounds as shown in Table 2 was taken from the paper by Ando et al. [9]. Ando’s experiments were performed with about 1–2 mg samples in an aluminum cell and a scanning rate of 10 °C min−1, for 820 reactive chemicals of which 18 nitro compounds were chosen in this work. These two data sets were treated separately to identify two correlations since it is very important to use values from the most similar conditions using as much as possible the same equipment. Onset temperature values of these nitro compounds were in the range from 223 to 379 °C.
Theoretical methods
Molecular simulation methods have been well established and extensively applied to study thermal hazards and thermal chemical/physical properties in our previous research [22–24]. In this work, all molecular structures were entered into and optimized using the Gaussian 03 suite of programs [25]. A density functional method, B3LYP (Becke 3 Lee, Yang, and Parr) [26, 27], was used for gas-phase geometry optimizations and frequency calculations at 298 K and 1 atm. The basis set 6-31+G(d) with Pople-style basis sets, including polarization functions for angular flexibility to represent regions of high electron density among bonded atoms was used [28]. Relative energies were reported in kcal mol−1 at the B3LYP/6-31+G(d) level of theory. Excitation energies can be easily calculated by simply taking the difference of the single point energy values for a singlet state and a triplet state. Correlations were then calculated based on singlet to triplet excitation energies and their experimental onset temperature values.
Results and discussion
As pointed out above, the rupture of C–NO2 bond in nitro compounds is a two-step process, while the first step is critical which needs more energy to excite electrons from a singlet state to a triplet state [19]. It is therefore possible that there is a simple relationship between excitation energy and onset temperature for nitro compounds.
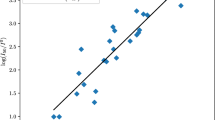
A correlation in Eq. (2) was developed based on the training set of 12 nitro compounds (see Table 1). Equation (2) shows the correlation between the onset temperature and the excitation energy with an R 2 value of 0.83. Figure 1 displays the onset temperature against the excitation energy.
Here T o is the onset temperature in °C and E x is the excitation energy in kcal mol−1. The second correlation was based on the experimental onset temperature data of 18 nitro compounds from another experiment setup, as shown below as Eq. (3).
Here T o is the onset temperature in °C and E x is the excitation energy in kcal mol−1. This correlation also has a good R 2 value of 0.79. The detailed information of the onset temperature against the excitation energy is displayed in Fig. 2. Figures 1 and 2 indicate that onset temperature and excitation energy are closely related to each other. The onset temperature can be approximately described as proportional to the excitation energy. The deviations are fairly small as shown in Tables 1 and 2. Those two correlations are theoretically justified and can provide significant insight into the relationship between the onset temperature of nitro compounds and their molecular structures.

A correlation between onset temperature and excitation energy based on data from the paper by Duh et al.

A correlation between onset temperature and excitation energy based on data from the paper by Ando et al.
Both Eqs. (2) and (3) are one-parameter correlations. Even though the maximum deviation could reach 16 °C for Eq. (2) and −40 °C for Eq. (3), the correlations provide pretty good estimations of the onset temperature. Previous work by Saraf et al. [18] assumed the activation energy was approximated as a fraction of the BDE, calculated for the C–NO2 bond. They then used those BDE values to predict onset temperatures for the similar series of nitro compounds. However, the deviation of the predicted onset temperatures could be as high as −96 °C [18]. Compared to the prediction results by Saraf et al., the predictions in this work are improved significantly with much smaller deviations.
It should be noted that a correlation model with two or more than two parameters may improve the accuracy of the prediction. A multiple linear regression (MLR) method should be used to describe the relationship among onset temperature and other parameters (number of parameters ≥2), such as C–N bond length and BDE.
It should also be noted that correlations reported in this work are limited to nitro aromatic compounds with only one NO2 functional group. Some nitro aromatic compounds with two NO2 functional groups (i.e., dinitrobenzene and dinitrotoluene) have been tested. The excitation energies were calculated using exactly same method and same basis set. However, there was no obvious relationship between the onset temperatures of those dinitro compounds and their excitation energies. There could be two reasons: (1) The overall excitation energies may shift due to the interaction of two NO2 functional groups on the same benzene ring; (2) As it is reported that the rupture of C–NO2 bond in nitro compounds is a two-step process, if the second step requires more energy and becomes the critical step, then the excitation energy may not be used to determine the decomposition rate and hence may not be related to the onset temperature.
Conclusions
The new correlations can be applied for calculating the onset temperatures of nitro aromatic compounds quickly. The predicted results of Eqs. (2) and (3) are reliable and can give better predictions as compared to outputs from the previous work which was correlated with BDE values. These correlations could be used as a computational screen tool to expedite the thermal hazard evaluation process of new developed nitro compounds.
References
Iwata Y, Momota M, Koseki H. Thermal risk evaluation of organic peroxide by automatic pressure tracking adiabatic calorimeter. J Therm Anal Calorim. 2006;85:617–22.
Wei C, Rogers WJ, Mannan MS. Layer of protection analysis for reactive chemical risk assessment. J Hazard Mater. 2008;159:19–24.
Wang Q, Rogers WJ, Mannan MS. Thermal risk assessment and ranking for reaction hazards in process safety. J Therm Anal Calorim. 2009;98:225–33.
Improving Reactive Hazard Management. U.S. Chemical Safety and Hazard Investigation Board Hazard Investigation, Washington, DC, 2002.
Olah GA, Spuire DR. Chemistry of energetic materials. San Diego: Academic Press; 1991.
Brill TB. Energetic materials: detonation, combustion. Amsterdam: Elsevier; 2003.
Keshavarz MH, Motamedoshariati H, Pouretedal HR, Tehrani MK, Semnani A. Prediction of shock sensitivity of explosives based on small scale gap test. J Hazard Mater. 2007;145:109–12.
Ding SD, Bai CY, Liu ZP, Wang YZ. Enhanced thermal stability of poly(p-dioxanone) in melt by adding an end-capping reagent. J Therm Anal Calorim. 2008;94:89–95.
Ando T, Fujimoto Y, Morisaki S. Analysis of differential scanning calorimetric data for reactive chemical. J Hazard Mater. 1991;28:251–80.
van Ekeren PJ, Bevers ERT. Temperature calibration of a high-pressure DSC for measurements in ammonia. J Therm Anal Calorim. 2007;90:931–4.
Chou YP, Hou HY, Chang RH, You ML, Peng JY, Shu CM. Thermal decomposition of cumene hydroperoxide in the presence of three incompatible substances by isothermal microcalorimetry and high performance liquid chromatography. J Therm Anal Calorim. 2009;96:771–5.
Dremin AN. Discoveries in detonation of molecular condensed explosives in the 20th century. Combust Explos Shock Waves. 2000;36:704–15.
Wu CJ, Fried LE. Ab initio study of RDX decomposition mechanisms. J Phys Chem A. 1997;101:8675–9.
Gindulyté A, Masaa L, Huang L, Karle J. Proposed mechanism of 1,1-diamino-dinitroethylene decomposition: a density functional theory study. J Phys Chem A. 1999;103:11045–51.
Alavi S, Reilly LM, Thompson DL. Theoretical predictions of the decomposition mechanisms of 1,3,3-trinitroazetidine (TNAZ). J Chem Phys. 2003;119:8297–304.
Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP. Advances in science and technology of modern energetic materials: an overview. J Hazard Mater. 2008;151:289–305.
Brill TB, James KJ. Kinetics and mechanisms of thermal decomposition of nitroaromatic explosives. Chem Rev. 1993;93:2667–92.
Saraf SR, Rogers WJ, Mannan MS. Application of transition state theory for thermal stability prediction. Ind Eng Chem Res. 2003;42:1341–6.
Wang Q, Ng D, Mannan MS. Study on the reaction mechanism and kinetics of the thermal decomposition of nitroethane. Ind Eng Chem Res. 2009;48:8745–51.
Manaa MR, Fried LE. DFT and ab initio study of the unimolecular dissociation of the lowest singlet and triplet states of nitromethane. J Phys Chem A. 1998;102:9884–9.
Duh YS, Lee C, Hsu CC, Hwang DR, Kao CS. Chemical incompatibility of nitrocompounds. J Hazard Mater. 1997;53:183–94.
Wang Q, Zhang Y, Rogers WJ, Mannan MS. Molecular simulation studies on chemical reactivity of methylcyclopentadiene. J Hazard Mater. 2009;165:141–7.
Wang Q, Wei C, Porez LM, Rogers WJ, Hall MB, Mannan MS. Thermal decomposition pathways of hydroxylamine: theoretical investigation on the initial steps. J Phys Chem A. 2010;114:9262–9.
Wang Q, Mannan MS. Prediction of thermochemical properties for gaseous ammonia oxide. J Chem Eng Data. 2010;55:5128–32.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004.
Becke AD. Density-functional thermochemistry. 3. The role of exact exchange. J Chem Phys. 1993;98:5648–52.
Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–9.
Frisch MJ, Pople JA, Binkley JS. Self-consistent molecular-orbital methods 25. Supplementary functions for Gaussian-basis sets. J Chem Phys. 1984;80:3265–9.
Acknowledgements
This research was supported by the Oklahoma State University Department of Fire Protection & Safety, and School of Chemical Engineering. We thank the supercomputing facility at Oklahoma State University for computer time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Q., Wang, J. & Larranaga, M.D. Simple relationship for predicting onset temperatures of nitro compounds in thermal explosions. J Therm Anal Calorim 111, 1033–1037 (2013). https://doi.org/10.1007/s10973-012-2377-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2377-4