
Abstract
Morphological and thermodynamic transitions in drugs as well as their amorphous and crystalline content in the solid state have been distinguished by thermal analytical techniques, which include dielectric analysis (DEA), differential scanning calorimetry (DSC), and macro-photomicrography. These techniques were used successfully to establish a structure versus property relationship with the United States Pharmacopeia standard set of active pharmaceutical ingredient (API) drugs. A distinguishing method is the DSC determination of the amorphous and crystalline content which is based on the fusion properties of the specific drug and its recrystallization. The DSC technique to determine the crystalline and amorphous content is based on a series of heat and cool cycles to evaluate the drugs ability to recrystallize. To enhance the amorphous portion, the API is heated above its melting temperature and cooled with liquid nitrogen to −120 °C (153 K). Alternatively a sample is program heated and cooled by DSC at a rate of 10 °C min−1. DEA measures the crystalline solid and amorphous liquid API electrical ionic conductivity. The DEA ionic conductivity is repeatable and differentiates the solid crystalline drug with a low conductivity level (10−2 pS cm−1) and a high conductivity level associated with the amorphous liquid (106 pS cm−1). The DSC sets the analytical transition temperature range from melting to recrystallization. However, analysis of the DEA ionic conductivity cycle establishes the quantitative amorphous and crystalline content in the solid state at frequencies of 0.10–1.00 Hz and to greater than 30 °C below the melting transition as the peak melting temperature. This describes the “activation energy method.” An Arrhenius plot, log ionic conductivity versus reciprocal temperature (K−1), of the pre-melt DEA transition yields frequency dependent activation energy (E a, J mol−1) for the complex charging in the solid state. The amorphous content is inversely proportional to the E a where the E a for the crystalline form is higher and lower for the amorphous form with a standard deviation of ±2%. There was a good agreement between the DSC crystalline melting, recrystallization, and the solid state DEA conductivity method with relevant microscopic evaluation. An alternate technique to determine amorphous and crystalline content has been established for the drugs of interest based on an obvious amorphous and crystalline state identified by macro-photomicrography and compared to the conductivity variations. This second “empirical method” correlates well with the “activation energy” method.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Riga et al. studied the extensive applications of dielectric analysis (DEA) including states of matter as amorphous/crystalline. They characterized drugs, excipients, transdermal patches, carbohydrates, proteins, amino acids, motor oil dispersants, and surfactants as well as electro rheological fluids using A.C. electrical properties over a wide range of frequencies [1]. The importance of the amorphous state when studying bioavailability, dissolution, and the development of poorly water soluble APIs has grown significantly over the recent years [2, 3]. There are two forms of solids: glassy and crystalline forms [4]. Glassy forms or amorphous solid forms generally exist in a variety of industrial fields or products, such as polymers and plastics, textiles, foods and pharmaceuticals, and in the manufacture of semiconductors, ceramics, metals, and optical materials [2, 3]. With reference to a crystalline solid, an amorphous solid can be defined as a substance with short-range molecular order; in contrast a crystalline solid has long-range order [2]. For this reason, some amorphous forms are considered as liquids although they can solidify by the removal of thermal energy or a solvent in a way that avoids crystallization [5]. In pharmaceutical research, the amorphous form of a pharmaceutical solid has been the most important aspect of drug development. The significance of amorphous solids is presently ever-increasing due to their value to the pre-clinical formulation scientists and in general to the pharmaceutical industry due to various advantages [3, 6, 7]: (i) a continuous increase in the development of a number of insoluble APIs; these glassy drug compounds are unique based on their methods of production and screening [8], (ii) the growing attention in regulatory evaluation and distinct economic aspects of pharmaceutical solids development [9], and (iii) the different polymorphs of APIs including the amorphous forms have different interconvertible physical and chemical properties [10] which exhibit different solubility and compressibility characteristics. Processing of an amorphous drug substance is relatively simple, and easy when compared to crystalline drug production [2, 3].
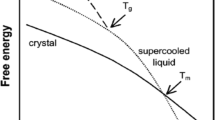
Amorphous forms can be produced by a variety of pharmaceutical techniques, such as granulation, compaction, freeze and spray drying, melt-quench cooling, and solvent evaporation method. These amorphous forms can be used for better solubility, dissolution, and bioavailability [6, 11, 12]. The amorphous phase has higher energy with higher chemical reactivity and generally is less stable both physically and chemically than that of the corresponding crystalline phase. The stability of the amorphous form is the main issue when stored at temperatures close to the glass transition temperature (T g). The glass transition temperature is a key characteristic property of amorphous materials. Quantification of the amorphous fraction of an API can be better understood by determining the heat capacity change associated with the T g [13]. In pharmaceuticals, amorphous solids have several useful properties and are used as both API and excipients [3]. Some of the useful properties are a higher water solubility, higher dissolution rate [14] (i.e., no lattice energy, which is a thermodynamic barrier to dissolution) [15], and better compression characteristics when compared to the crystalline forms [2, 3].
Although the amorphous form has some advantages over its crystalline counterpart, like higher free energy, lattice disorder, higher water solubility, and molecular mobility [6], the amorphous systems have seen limited commercialization due to its thermodynamic instability and higher chemical reactivity [11, 12, 16]. The amorphous phase can also occur throughout the particle or solid, in the bulk or at its surface, which can be relatively distinguished by dielectric studies [17]. As the disordered portion is low, it is difficult to detect [11, 12]. But these high-energy reactive portions may cause some significant changes in drug development like: improve product performance, high dissolution rate, decreased chemical stability, solid–solid transitions, and recrystallization during storage [11, 18]. Because of the amorphous phase thermodynamic instability, there can be an irreversible conversion of the metastable form to its stable crystal form during manufacturing or in normal storage. Loss of drug function can occur when the amorphous material transitions to another metastable form resulting in, for example, a varying dissolution rate of the original crystalline form [16].
Therefore, one must find a reliable method to monitor and characterize the degree of crystallinity and the amount of disorder in the APIs during their pre-clinical drug development to insure commercializable formulations [11]. To maximize the efficiency of the amorphous material and prevent transformation of the drug during storage, one needs to understand and characterize the kinetics of the crystallization process, and determine quantitatively the various parameters associated with nucleation and crystal growth [16]. This need is based on a vigorous test method to monitor the drug throughout its shelf life to assure product safety and quality [18]. Pharmaceutical scientists are making an effort to develop alternative methods that are precise, reliable, and fast, which employ small samples for determining the crystallinity or low levels of amorphicity in the sample [12]. The degree of crystallinity methodology is extensively used to measure the relative crystalline/amorphous content in pharmaceutical materials [19]. Several analytical techniques currently employed in the pharmaceutical industry for quantifying, and/or characterization of crystalline and amorphous forms of pharmaceutical materials include: X-ray diffraction analysis, DEA [20], solid state-NMR [21], FT-Raman [22], FT-IR [23], modulated differential scanning calorimetry [24], thermally stimulated current spectroscopy [11], isothermal microcalorimetry, and solution calorimetry.
Morphological and thermodynamic transitions in drugs as well as their amorphous and crystalline content in the solid state have been initiated by thermal analytical techniques, which include DEA and thermal mechanical analysis [24, 25]. These techniques were used successfully to establish a structure versus property relationship with the API of drugs [24, 25]. The main focus of this study is to develop a novel analytical protocol utilizing DEA to define and quantify the crystalline and amorphous content in pharmaceutical solids. These innovative techniques will give pharmaceutical scientists new insights in how to understand the nature and behavior of drugs in the solid state. Pharmaceutical solids studied by these methods have led to a better understanding of the chemistry and molecular mobility that relates to the structure of the drug. This information is of the utmost importance during pre-formulation studies of drug development. DEA measures the crystalline solid and amorphous liquid API electrical conductivity at temperatures below and above the melting temperature [13–15]. DEA scans the electrical signals over a wide range of frequencies (e.g., 0.10–100,000 Hz). The DEA ionic-dielectric conductivity is repeatable and differentiates the solid crystalline drug with a low ionic-dielectric conductivity level (<10−1 pS cm−1) and a high ionic-dielectric conductivity level associated with the amorphous liquid (>105 pS cm−1). Further, DEA measures changes in phase (solid to liquid) transitions, significant variations in conductivity related to the ionic-dielectric behavior of melted drugs and loss of residual solvents as it is subjected to a periodic electric field.
Our new DEA protocols can detect the crystalline and amorphous phase repeatedly and rapidly based on the experimental frequencies, time and temperatures. This A.C. frequency based technique has the ability to differentiate surface versus bulk amorphicity [26]. Typically surface analysis is best profiled at 0.10–1.0 Hz and bulk analysis is between 1,000 and 10,000 Hz. This methodology is quick and can summarize the relative low levels of non-crystalline and crystalline material, thereby increasing the sensitivity of this technique.
Dielectric analysis (DEA)
DEA is a material characterization technique that provides scientists with quantitative thermal, rheological, and dielectric information on a wide range of materials in their various forms, which include solid, liquids, pastes, films, polymers, and organic additives in lubricating oils [16]. It can be used to determine the flow, thermal transitions, degree and rate of cure in polymers, characterize food products, pharmaceutical materials, and dielectric properties of thermoplastics, composites, as well as adhesives and coatings [1].
As DEA is a thermal analysis tool, it is closely related to DMA (frequency dependent dynamic mechanical analysis), which measures mechanical properties of a material. While differential scanning calorimetry (DSC) measures heat flow through which the heat of fusion and transition temperatures can be determined. DEA compliments DSC by allowing a measurement of molecular motion based on its electrical properties.
The theory of a dielectric or theory of DEA may be illustrated by the time-dependent electrical response of a sample placed on a single surface gold ceramic interdigitated electrode when an alternating (sinusoidal voltage) electric field is applied. This process produces polarization within the sample, causing oscillation at the same frequency as the electric field but with a phase angle (or phase shift) δ. This phase angle shift can be measured by comparing the applied voltage to the measured current [1, 24]. The current is then separated into capacitive (e′) and conductive (e″) components.
The two fundamental characteristics of a material measured through dielectrics: as a function of time, temperature, and sampling frequency are:
-
Capacitance = High frequency permittivity (e′) or dielectric constant.
-
Electrical conductivity (pS cm−1) = Loss factor (e″) × Applied frequency (Hz) × 2π
The capacitive nature of the material allows or has an ability to store an electrical charge and this nature dominates the electrical response at low temperatures. The conductive nature of the material has an ability to transfer an electric charge and this factor becomes very important when the solid material is heated above its melting temperature to the liquid state. These electrical properties are significant since they have been related to molecular activity, allowing for probing the chemistry, and molecular mobility of polymers and pharmaceutical materials. DEA reports three main electrical signals over a wide range of frequencies (e.g., 0.10–100,000 Hz):
-
Permittivity (e′) is a measure of the induced dipoles and alignment of molecular groups (dipoles) in the electric field.
-
Loss factor (e″) is a measure of the energy required to move the molecular groups or ions and is proportional to ion conductivity. Ionic conductivity is associated with the viscosity of the sample because fluidity is identified by the ease with which ionic components can migrate through the sample under the applied electric field.
-
Tan delta is the ratio of the loss factor divided by the permittivity. Tan delta = e″/e′. Tan delta values are related to molecular mobility, response time to an electric field, and are related to polarization or relaxation of excited molecules or a measure of charge transfer properties [1, 24].
We developed a novel combined DEA–DSC protocol to evaluate amorphous/crystalline content in pharmaceutical solids. The DSC analysis will be used in two different protocols: (a) heat cycle only to measure the temperature range for heating to aid DEA evaluation, and (b) heat and Cool cycle to evaluate % crystalline and % amorphous based on the fusion and crystallization heats.
The focus of these new methods is to determine the relative amounts of amorphous and crystalline content in APIs. Pharmaceutical scientists working on pre-formulation of pharmaceutical drugs as well as those developing quality control of manufactured drugs need new information on the content of amorphous/crystalline matter. This new knowledge is to improve quality and stability of pharmaceuticals for the consumer. There is a clear rationale for developing these new methods to better understand the effects of the total chemical structure on drug treatment including amorphicity. It is logical that electrical conductivity methods are implemented now since we have discovered significant variations in pharmaceuticals using DEA.
Methodology
Two novel DEA protocols were developed to determining the amorphous and crystalline content in pharmaceutical drugs. The first method is “The empirical method.” It is semiquantitative method that is based on the observed significant electrical conductivity difference between the amorphous liquid (107 pS cm−1) and crystalline solid (10−2 pS cm−1). The second method is the “activation energy method” and is more quantitative in nature. In this method, the activation energy, E a (J mol−1), is calculated from the DEA ionic conductivity. It is based on the fact that the relative E a for the electrical reactivity (charging or charge transport) in the solid state of the amorphous phase is higher and conversely the E a is lower (typically 30–110 J mol−1) and the electrical reactivity of the solid state for crystalline phase is low and conversely the E a is higher (typically 1,200–2,000 J mol−1).
Empirical method protocol
The empirical method to determine crystalline and amorphous content in pharmaceutical solids is as follows: First, a pure drug (100% crystalline) similar to the drug with unknown content is examined by DEA as a standard. For the pure drug prepared by the pharmaceutical company the % content before the melting temperature is 99.99–100% crystalline and with complete melt it is 100% amorphous in the liquid phase. Then, DEA re-run of the sample is performed to analyze the drug with the unknown amorphous/crystalline content. A combined DEA model plot for the % content of an unknown drug and the pure drug is shown in the graphical representation (Fig. 1) as log ionic conductivity versus temperature. Next, a temperature, T s, is selected ≤30–50 °C below the melting temperature (T m) of the drug from the overlayed DSC curve. Then, the linear distance (Δ3) in millimeters of log ionic conductivity on the y-axis is measured from 100% solid crystalline line (point X) to the 100% amorphous line (point Z) at the selected temperature T s. The 100% amorphous or liquid region is a hypothetical region and the 100% amorphous line is obtained by extrapolating the liquid region log ionic conductivity of the DEA plot. Next, the linear distance (Δ2) in millimeters of log ionic conductivity on the y- axis is measured from the semicrystalline line (point Y) to the 100% amorphous line (point Z) and (Δ1) in millimeters from 100% solid crystalline line (point X) to semicrystalline line (point Y) at the temperature T s are measured. In the semicrystalline region, the ratio of amorphous to crystalline content is not known.

Graphical representation of DEA empirical method
The linear log conductivity distance in millimeters is a measure from the crystalline phase to the amorphous phase, which is equated to 100%. The Amorphous and crystalline contents can be calculated from the following equations and Fig. 1.
Activation energy method
A second method was developed based on a measurement of a DEA activation energy E a (J mol−1) to determine the amorphous and crystalline content in drugs by DEA. Sharma and Yashonath [27] investigated ionic transport in a variety of inorganic amorphous glasses (solids). They observed a strong correlation between ionic conductivity and activation energy. Further, they found that higher conductivity is associated with lower activation energies and lower conductivity is associated with higher activation energies. Their results suggest that there is a strong relation between microscopic structure of the amorphous solid, ionic conductivity, and activation energy [27].
The E a (J mol−1) calculation is more quantitative and is based on the relative activation energy for the electrical reactivity (charging or charge transport) in the solid state. The amorphous phase reactivity is higher (ca. 107 pS cm−1) and conversely the Ea is lower (frequency at 1 or 5 Hz typically: 60–200 J mol−1) measured as the slope of the log ionic conductivity versus reciprocal temperature (K−1) of an Arrhenius plot and as a function of frequency (Hz). The reactivity and conductivity (typically 10−2 pS cm−1) of the solid state crystalline phase is lower and conversely the Ea is higher (typically measured by DEA at a frequency of 1 or 5 Hz: 1,200–2,000 J mol−1), also measured by DEA conductivity versus reciprocal temperature (K−1) [28–30].
Activation energy E a (J mol−1) protocol
-
1.
Record a DEA curve of a pharmaceutical solid sample at a heating rate of 10°C min−1 in a nitrogen flow of 60 mL min−1; a sample size of 20 mg. Heat to 20–30 °C above the peak melting temperature recorded in the DSC curve.
-
2.
Plot the Ionic conductivity, pmho cm−1 versus Temperature at fixed frequencies of 0.10–1.0 Hz.
-
3.
Measure the Ionic conductivity responses at <30 °C below the DSC melting point temperature.
-
4.
From the measured responses plot an Arrhenius plot of DEA log ionic conductivity versus (K−1) × 1000 and perform a linear curve fit to determine the equation of the line. The slope of the linear curve fit is E a R −1and must be multiplied by 19.14, to convert the slope value into E a (J mol−1). Note: E a is the activation energy and R is the gas constant of 8.314 J K−1 mol−1 and one must include the conversion of natural log (ln) to log base 10 which is 2.303.
-
5.
The E a (J mol−1) is inversely proportional to the concentration of the amorphous phase in the pharmaceutical sample. Relatively, the lower the Ea corresponds to a higher the ionic conductivity. The first DEA run resulting in log conductivity versus Temperature yielding % crystallinity based on the DSC purity by heat of fusion (J g−1). For an API, the % crystallinity is probably 100% for the first sampling by DEA. For the second and third runs, samplings of the same API it will be less crystalline and more amorphous, based on a lower E a (J mol−1) value.
Experimental
Drugs
The following drugs were evaluated in this study: Lidocaine (T m, 68 °C), Lidocaine·HCl (T m, 74–79 °C), Sulfapyridine (T m, 192 °C), Indomethacin (T m, 155 °C), and Acetophenetidin (T m, 135 °C). All the drugs were obtained from Sigma-Aldrich® and meet the USP (United State Pharmacopeias) testing specifications. Quetiapine fumarate (Seroquel®) (T m, 173 °C), and Procainamide·HCl (T m, 169 °C) were purchased from Haorui Pharma-chem Inc. Edison, NJ.
Experimental procedure
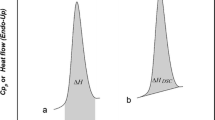
A TAI 2920 DSC (TA Instrument) was used to profile the drug solid–solid transitions, melting, glass transition (T g), and crystallization properties of the drugs. All the samples were ramped at a rate of 10 °C min−1 in purged nitrogen gas flow at a rate of 60 mL min−1 during heating and cooling cycles. The DSC aluminum pan weighed 13–14 mg. Samples in the range of 8–10 mg were weighed using the Mettler AT261 Delta Range® microbalance and loaded into an aluminum pan then closed and crimped. Temperature range was typically from room temperature to 30 °C above the peak melting temperature (T m) for the drugs under study. Heat flow (W g−1) values versus temperature and time were generated using the (TA Thermal Advantage) Universal Analysis 2000 software. The DSC instrument was calibrated using Indium as a standard according to ASTM standard test protocol E967.
A TAI 2970 DEA (TA Instrument) was used to determine the electrical conductivity and Tan delta curve for each drug studied. For each solid powdered drug, a sample of approximately 20 mg was placed on a single surface gold ceramic interdigitated sensor. The samples were ramped from room temperature to 30 °C above the melting temperature of the drug at heating rate of 10 °C min−1. A purge gas of nitrogen gas flowed at the rate of 60 mL min−1. The gold ceramic interdigitated sensors were calibrated by the fixture supplied by TAI. The DEA instrument was further calibrated according to ASTM E2038 Standard Test Method for Temperature Calibration of Dielectric Analyzers. This standard method was employed for all the drugs studied at 5 and 10 °C min−1. DEA was used to evaluate the electrical properties of the drugs. The conductivity measurements were recorded at controlled interval frequencies ranging from 0.10 to 10,000 Hz for all temperatures.
A Konica-Minolta DG SLR Camera with a 28–80 mm macro-lens was used for macro-photomicrography to characterize the crystalline and amorphous materials on the single surface interdigitated array gold electrodes, and samples on the DSC pans. An eye piece reticule was used to calibrate the macro-photomicrography system.
Results and discussions
We tested the applicability of the novel empirical method with several model pharmaceutical APIs. When Lidocaine is heat cycled through the melt and then cooled back to room temperature. The first DEA run of Lidocaine reveals the crystalline to amorphous phase transition and change in the ionic-dielectric conductivity from ≤10−2 (pS cm−1) for the crystalline phase and 105 (pS cm−1) for the amorphous phase. Figure 2 shows the DEA surface analysis profile for Lidocaine at 0.5 Hz. Low frequency DEA is confirmed as measurements at the electrode surface, e.g., 0.5 Hz. The Lidocaine (Sigma-Aldrich®) sample is initially 99.99% crystalline with very low ionic conductivity for the first run and upon cooling and reheating a huge increase in ionic conductivity was observed. The second and third runs commensurate with an increasing ionic conductivity value of 103 (pS cm−1) (79% amorphous) and 105 (pS cm−1) (91% amorphous) (see Figs. 2, 3). Figure 3 summarizes the % crystalline and % amorphous content in Lidocaine at 0.1, 0.5, and 1.0 Hz by the DEA “Empirical” method. Figure 4 summarizes the % crystalline and % amorphous content in Quetiapine Fumarate (Seroquel®) at 0.1, 0.5, and 1.0 Hz by the DEA empirical method. Table 1 shows the list of drugs tested and evaluated for crystalline and amorphous content by the proposed method (Table 1).

DEA Surface Analysis of Lidocaine at 0.5 Hz (first, second, third runs); DSC T m 69.89 °C

Determination of % crystalline and % amorphous content versus Frequency/Hz for Lidocaine at 0.1, 0.5, and 1.0 Hz (runs 1–3) by the DEA empirical method. % amp % amorphous content, % cry % crystalline content

Determination of % crystalline and % amorphous content versus Frequency/Hz for Quetiapine Fumarate at 0.1, 0.5, and 1.0 Hz (runs 1–3) by the DEA empirical method. % amp % amorphous content, % cry % crystalline content
Our second method to determine crystalline and amorphous content through the measurement of DEA activation energies was tested with several model pharmaceutical APIs. Results for Lidocaine and Acetophenetidin are discussed as follows. Analysis of the Lidocaine by the proposed DEA empirical method yields a supportive interpretation of the activation energy method, i.e., determination of the % crystalline and amorphous content in a drug. Other APIs successfully tested by the activation energy method are Sulfapyridine, Quetiapine Fumarate (Seroquel®), Procainamide·HCl, Lidocaine·HCl, and Indomethacin.
A plot of the DEA Log Ionic conductivity (pS cm−1) versus Temperature (°C) for crystalline Lidocaine is summarized in Fig. 5. For the first run, we observed a slow rise in ionic conductivity till 60 °C, (10−2 pS cm−1) followed by a very rapid increase (105 pS cm−1) from 60 to 90 °C. The DSC curve overlaid on the DEA plot maps out the melting transition that occurs at 67 °C. The second run cycling after the initial heat curve produced ionic conductivity response that was enhanced due to the formation of the amorphous phase in Lidocaine, was observed from the temperature range of 40–70 °C. The slope of the first curve was steep and decreased significantly in the second and third run. The slope change tracked the decreasing activation energy with each heat cycle. The ionic conductivity increases proportionally with the enhanced amorphous content. A curve fit of the log ionic conductivity versus the reciprocal temperature in Kelvin produces a linear curve with a correlation coefficient of R 2 = 0.999 and a varying activation energy, for Lidocaine (Run 3) the E a is 27 J mol−1 (see Fig. 6) for details. Table 2 summarized the Lidocaine data in triplicate for the DEA activation energy method and the determination of the % crystalline and amorphous content produced by a heat cycle at three frequencies, 0.1, 0.5, and 1.0 Hz (all frequencies are associated with occurrences at the sensor surface). The second and third runs produced an amorphous content of 93% at 0.1 Hz, 94% at 0.5 Hz, and 1.0 Hz (see Table 2).

Lidocaine DEA and DSC curve overlay and comparing ionic conductivity in crystalline and amorphous samples by activation energy method at 0.1–1.0 Hz (runs 1–3); T m = 69.89 °C

Log conductivity (k) versus 1/T (Kelvin) × 1000 for Lidocaine (run 3, 0.1 Hz); E a = 27 J mol−1
Evaluation of Acetophenetidin by the proposed DEA method for amorphous–crystalline content was an outlier in this protocol, since this drug recrystallizes significantly while the other six drugs did not recrystallize to a noticeable extent. The activation energy for Acetophenetidin described in (Figs. 7, 8) yielded a high value of 1,240 J mol−1 at low ionic conductivities. It is our interpretation that this drug continues to recrystallize and therefore the method to quantify the concentration ratio of crystalline to amorphous varies. The second and third run produced varying values for the % amorphous and % crystalline content with frequency. The content was frequency dependent for this drug but none of the other drugs evaluated. The second and third runs produced an amorphous content of 25, 49% for 0.1 Hz; 58, 70% for 0.5 Hz; and 46, 69% for 1.0 Hz, respectively.

Acetophenetidin DEA and DSC curve overlay and comparing Ionic conductivity in crystalline and amorphous samples by activation energy method at 0.1–1.0 Hz (runs 1–3); T m = 135.80 °C

Log conductivity (k) versus 1/T (Kelvin) × 1000 for Acetophenetidin (run 1, 0.1 Hz); E a = 1,244 J mol−1
Quantification of the amorphous and crystalline content of the drugs studied by the DEA activation energy method is summarized in Table 3. A correlation was established between the empirical method and activation energy method resulting in the correlation coefficient of R 2 = 0.925 for the overall average % amorphous content of all the drugs studied (see Fig. 9).

A correlation graph of empirical method versus activation energy method (E a) for the overall average % amorphous content of all the drugs studied
Semiqualitative evaluation of the amorphous and crystalline states by macro-photomicrography clearly denotes the two phases (see Fig. 10) for Sulfapyridine at 2.4× magnification. See Fig. 11 for amorphous melt of Lidocaine·HCl after third run. All Pharmaceutical solids used in this study were characterized by this method. This material characterization technique aids the development of the DEA methods for identifying amorphous and crystalline character of the drug.

Macro-photograph of DEA electrodes with sulfa pyridine in crystalline powder form (right), and amorphous form (left) at ×2.4 magnification

Macro-photograph of DEA electrode with amorphous melt of Lidocaine·HCl after third run
Conclusions
The most prominent conclusion is that we now have quantitative methods for determining the crystalline and amorphous content of pharmaceutical solids by DEA. We are basing this conclusion on the facts that the API’s distributed by pharmaceutical companies are ≥99.99% pure crystalline solids. Once the crystalline state is completely lost during melting the material becomes 100% amorphous.
Our studies reveal that six out of the seven drugs evaluated showed this behavior of complete amorphous formation. In the case of Acetophenetidin, it recrystallized rapidly while the others remained predominantly amorphous. The crystalline and amorphous content of the drugs as determined by the empirical method was frequency independent. The frequencies studied relate to surface analysis at 0.1–1.0 Hz. The crystalline and amorphous content of the drugs as determined by the activation energy method was frequency dependent for one drug, i.e., Acetophenetidin. Therefore, Acetophenetidin was an outlier for these methods. DEA for the first run of the API’s is 100% crystalline with lower ionic conductivity and high activation energy, e.g., Acetophenetidin 1,200–1,800 J mol−1. The second and third run of this drug showed repeatable variations with higher ionic conductivity and lower activation energy observed from 470 to 930 J mol−1. The other drugs investigated yielded a stable activation energy, e.g., for Lidocaine crystalline form it was 480 J mol−1 initially and for the amorphous form it was 27 J mol−1 at 1.0 Hz.
A comparison of overall average amorphous content by the empirical method had a linear relationship with the activation energy method with a correlation coefficient of R 2 = 0.925. The correlation coefficient for overall average crystalline content was R 2 = 0.94. After calibrating the DEA instrument, examining a 10-mg sample with a heating and cooling profile plus interpretation time will lead to a definitive amorphous content in 1 h. Marco-photomicrography aids the DEA interpretation as an opaque solid material as crystalline solid against the transparent amorphous material. These methods determine the % amorphicity and % crystallinity and are significant contributions to the analytical science of pharmaceuticals.
References
Riga A, Judovits L. Materials characterization by dynamic and modulated thermal analytical techniques. West Conshohocken: ASTM. Special technical publication; 2011.
Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev. 2001;48:27–42.
Jójárt-Laczkovich O, Szabó-Révész P. Amorphization of a crystalline active pharmaceutical ingredient and thermo analytical measurements on this glassy form. J Therm Anal Calorim. 2009. doi:10.1007/s10973-009-0530-5.
Cui Y. A material science perspective of pharmaceutical solids. Int J Pharm. 2007;339:3–18.
Hancock BC. Disordered drug delivery: destiny, dynamics and the Deborah number. J Pharm Pharmacol. 2002;54:737–46.
Hancock BC, Zografi G. Characteristic and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86:1–12.
Keré J, Sréié S. Thermal analysis of glassy pharmaceuticals. Thermochim Acta. 1995;248:81–95.
Angell CA. The old problems of glass and the glass transition, and the many new twists. Proc Natl Acad Sci USA. 1995;92:6675–82.
Byrn S, Pfeiffer R, Ganey M, Hoi berg C, Poochkian G. Pharmaceutical solids: a strategic approach to regulatory consideration. Pharm Res. 1995;12:945–54.
Vippagunta Sr, Brittain HG, Grant DJW. Crystalline solids. Adv Drug Deliv Rev. 2001;48:3–26.
Venkatesh GM, Barnett ME, Fordjour CO, Galop M. Detection of low levels of the amorphous phase in the crystalline materials by thermally stimulated current spectrometry. Pharm Res. 2001;18:98–103.
Shah B, Kaumanu VK, Bansal AK. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J Pharm Sci. 2006;95:1641–65.
Lappalainen Minna, Karppinen Maarit. Techniques of differential scanning calorimetry for quantification of low contents of amorphous phases. J Therm Anal Calorim. 2010;102:171–80.
Hancock BC, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17:397–403.
Singhal D, Curatolo W. Drug polymorphism and dosage form design: a practical perspective. Adv Drug Deliv Rev. 2004;56:335–47.
Schmitt EA, Law D, Zhang GGZ. Nucleation and crystallization kinetics of hydrated amorphous lactose above the glass transition temperature. J Pharm Sci. 1999;88:291–6.
Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperature. Pharm Res. 1995;12:799–806.
Seyer JJ, Luner PE, Kemper MS. Application of diffuse reflectance near infrared spectroscopy for the determination of crystallinity. J Pharm Sci. 2000;89:1305–16.
Hendrickson BA. Characterization of calcium fenoprofen 1. Powder dissolution rate and degree of crystallinity. Int J Pharm. 1990;60:243–52.
Duddu SP, Sokoloski TD. Dielectric analysis in the characterization of amorphous pharmaceutical solids. 1. Molecular mobility in poly (vinylpyrrolidine)–water systems in the glassy state. J Pharm Sci. 1995;84:773–6.
Gustafson C, Lennholm H, Iversen T, Nystrom C. Comparison of solid state NMR and isothermal microcalorimetry in the assessment of the amorphous component of lactose. Int J Pharm. 1998;174:243–52.
Taylor LS, Zografi G. The quantitative analysis of crystallinity using FT-Raman spectroscopy. Pharm Res. 1998;15:755–61.
Black DB, Lovering EG. Estimation of the degree of crystallinity in digoxin by X-ray and Infrared methods. J Pharm Sci. 1997;29:684–7.
Riga Alan T, Alexander Kenneth S. Electrical conductivity analysis/dielectric analysis differentiates physical-chemical properties of drugs and excipients. Am Pharm Rev. 2005;8(6):45–51.
Thakur SS, Maheswaram MPK, Mantheni DR, Kaza L, Perera I, Ball DW, Moran J, Riga AT. Solid–state mechanical properties of crystalline drugs and excipients: new data substantiate discovered dielectric viscoelastic characteristics. J Therm Anal Calorim. 2011. doi:10.1007/s10973-011-18559-0.
Mantheni DR, Maheswaram MPK, Sobhi HF, Perera NI, Riga AT, Ellen Matthews M, Alexander K. Solid state studies of drugs and chemicals by dielectric and calorimetric analysis. J Therm Anal Calorim. 2011. doi:10.1007/s10973-011-1423-y.
Sharma M, Yashonath S. Correlation between conductivity and diffusivity and activation energy in amorphous solids. J Chem Phys. 2008;129:144103.
Matthews ME, Atkinson I, Presswala L, Najjar O, Gerhardstein N, Wei R, Rye E, Riga AT. Dielectric classification of d- and l-amino acids by thermal and analytical methods. J Therm Anal Calorim. 2008;93:281–7.
Presswala L, Matthews ME, Atkinson I, Najjar O, Gerhardstein N, Moran J, Wei R, Riga AT. The discovery of bound and unbound waters in crystalline amino acids revealed by thermal analysis. J Therm Anal Calorim. 2008;93:295–300.
Matthews ME, Riga AT. Effects of thermal history on solid state and melting behavior of amino acids. J Therm Anal Calorim. 2009;96(3):673–6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maheswaram, M.P., Mantheni, D., Perera, I. et al. Characterization of crystalline and amorphous content in pharmaceutical solids by dielectric thermal analysis. J Therm Anal Calorim 111, 1987–1997 (2013). https://doi.org/10.1007/s10973-011-2140-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-011-2140-2