
Abstract
Ibuprofen (C15H18O2) is an anti-inflammatory drug. It is important to investigate its structure to know the active groups and weak bond responsible for its medical activity. Consequently in the present study, ibuprofen was investigated by mass spectrometry (MS) and thermal analyses (TAs) (TG/DTG and DTA), and confirmed by semi-empirical molecular orbital (MO) calculation using PM3 procedure, on the neutral and positively charged forms of the drug. These calculations included bond order, bond length, and bond strain, and charge distribution, heat of formation, and ionization energy. The mass spectra and thermal analysis fragmentation pathways were proposed and compared to each other to select the most suitable scheme representing the correct fragmentation pathway of the drug in both techniques. From the electron ionization (EI) mass spectra, the primary cleavage site of the charged molecule is because of the rupture of COOH group (the lowest bond order) followed by propyl group loss. The TAs of the drug revealed high response of the drug to the temperature variation with very fast rate. It decomposed in several sequential steps in the temperature range 25–360 °C. The initial thermal decomposition is similar to that obtained by MS fragmentation of the first rupture (COOH), then subsequent one of propyl loss, and finally of ethylene loss. These mass losses appear as endothermic peaks required energy values of −214.83, −895.95, and −211.10 J g−1, respectively. The order of these losses is also related to the values of the MO calculation parameters. Therefore, the comparison between MS and TA helps in the selection of the proper pathway representing the decomposition of this drug to give its metabolites in in vivo system. This comparison is also successfully confirmed by MO calculations.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ibuprofen (IB, C15H18O2, MW = 206) drug, has an IUPAC name (R.S.-2-4-isobutylphenyl) propionic acid, as a derivative of 2-arylpropionic acid. It is a chiral non-steroidal anti-inflammatory drug [1]. It is used for relieving moderate pain, acute arthritis, non-rheumatic inflammation, fever, and dysmenorrheal [2]. The structure and the proposed numbering system of the drug are shown in Fig. 1.

Normal structure of ibuprofen and numbering system
Rapid advances in biological sciences have led to an increased demand for the chemical and structural information about biological systems. The mass spectrometry (MS) plays a vital role in the structural characterization of biological molecules [3]. The technique is important because it provides a lot of structural information with little expenditure of the sample. Also, the technique offers comparative advantages of speed and productivity for pharmaceutical analysis [4]. On the other hand, thermal analysis technique that delivers extremely sensitive measurements of heat changes can be applied on broad scale with pharmaceutical development. These methods provide unique information in relation to thermodynamic data of the system studies. The increasing use of the combined techniques is providing more specific information, and thus facilitates rapid interpretation of the experimental curves obtained [5].
Electron ionization mass spectral (EI-MS) fragmentation consists of competitive and consecutive unimolecular fragmentation pathways [6]. The fragmentation of ionized molecule depends mainly on their internal energy [7]. The thermogravimetric (TG/DTG) analysis has been employed to provide quantitative information on mass losses due to the decomposition and/or evaporation of low molecular materials as a function of time and temperature. In conjunction with mass spectrometric analysis [8–10], the nature of the released volatilize fragment may be deduced, thus greatly facilitating the interpretation of thermal degradation processes. On the other hand, computational quantum chemistry can provide additional information about the atoms and bond, which can be used successfully in an interpretation of experimental results [11]. These additional computational data, also, can be used in the description and prediction of primary fragmentation site and subsequent ones. Detailed studies were carried out by Zayed et al. [12–15]. Also, there are a number of methods described in the literature for IB drug. These included direct and indirect liquid chromatographic methods [16], supercritical-fluid direct chromatography [17], and a combination of micellar electrokinetic chromatographic separation with mass spectrometric detection [18], capillary electrophoresis [19], diffused reflectance and FT-IR spectroscopy [20], and gas chromatography [21].
The aim of the present study is to focus on further application of our previous study [12–15] on ibuprofen drug. This study includes a correlation between EI-MS fragmentation and thermal analysis (TA) degradation of the drug, and comparison of the experimental data with the theoretical molecular orbital (MO) calculation to identify the weakest bonds ruptured during both mass and thermal studies. Consequently, the choice of the correct pathway of such fragmentation knowing this structural session of bonds can be used for deciding the active sites of this drug responsible for its chemical, biological, and medical reactivities.
Experimental
Mass spectrometry (MS)
The EI-MS of ibuprofen were obtained using Shimadzu GC-MS-Qp 1000 PX quadruple mass spectrometer with electron multiplier detector equipped with GC-MS data system. The direct probe (DP) for solid material was used in this study. The sample was put into a glass sample micro vial, by a needle (≈1 μg max), with the vial being installed on the tip of the DP containing heating cable and inserted into the evacuated ion source. The sample was ionized by electron beam emitted from the filament, with the generated ions being effectively introduced into the analyzer by the focusing and extractor lenses system. The MS was continuously scanned, and the obtained spectra were stored. The EI-MS were obtained at an ionizing energy value of 70 eV, an ionization current of 60 μA, with vacuum being better than 10−6 torr.
Thermal analyses (TAs)
The TAs of ibuprofen drug were made using conventional thermal analyzer (Shimadzu system of DTA-50 and 30 series TG-50). The mass losses of 5 mg sample and heat response of the change of the sample were measured from 25 up to 400 °C. The heating rate, in an inert argon atmosphere, was 10 °C min−1. These instruments were calibrated using indium metal as a thermal stable material. The reproducibility of the instrument reading was determined by repeating each experiment more than twice.
Computational method
The theoretical calculations were performed using semi-empirical MO calculation. The method used in these computations is the parametric method (PM3 method) described by Stewart [22]. The default criteria for terminating all optimizations were increased by a factor of 100 (keyword PRECISE). Vibration frequencies were computed for the studied structures (keyword FORCE) so as to check whether the newly designed geometries are local minima. All the MO calculations were carried out at the restricted Hartree–Fock level (RHF) for the neutral molecule of ibuprofen, while those at the unrestricted Hartree–Fock level (UHF) were carried out for its cation by means of PM-3 method followed by full optimization of all geometrical variables (bond lengths, bond angles, and dihedral angles), without any symmetry constraint. All the structures were optimized to a gradient norm range of 0.01–0.05, using the eigenvector-following (EF) routine [23]. All the semi-empirical MO calculations were performed using the MOPAC2000 software package [24] implemented on an Intel Pentium IV 3.0 GHz computer.
Results and discussion
The fragmentation in both EI-MS and TA techniques are similar where the rupture takes place at the weakest bond positions [6]. In TA, the molecules are continuously energized and deactivated by a gas evolution and the distribution of energy can be described as a function of temperature. In EI-MS, the ion produced is formed with a specific amount of internal energy, which is conserved independently in all subsequent dissociations. During ionization process, transitions to various states are possible leading to a collection of ions with a distribution of internal energies. The dissociation does not immediately follow ionization, but it is slow enough to permit the transfer of energy into the various degrees of freedom involved in the observed dissociation.
Thermogravimetric (TG/DTG) analysis is employed to provide quantitative information on weight losses due to decomposition and/or evaporation of low molecular material as a function of time and temperature. The particular problem in studying TA of large molecule is that the decomposition takes place in a series of overlapping reactions with multiple products. In conjunction with mass spectrometric analysis [8–10], the nature of the released volatile may be deduced, thus greatly facilitating the interpretation of thermal degradation processes.
The MO calculations give valuable information about the structure of the molecules [6], which may be used for supporting experimental evidence.
It is of great interest to study the chemistry and reactivity of ibuprofen drug because of its importance in medicine. Knowledge obtained from thermal decomposition mechanisms of the neutral drug is very important to understand the chemical process that is shared in biological systems. It is difficult to establish the exact major fragmentation pathway in EI using conventional MS. With the combination of the above two techniques and the data obtained from the MO calculation, it is possible to understand the following topics:
-
1.
Stability of the drug under thermal degradation in solid state and EI-MS fragmentation in gas phase.
-
2.
Prediction of the primary site of fragmentation and subsequent bond cleavage.
-
3.
The correct pathway in both techniques.
-
4.
Understanding what actually happens in biodegradation of the drug or its derivatives in vivo system and metabolites.
Mass spectrum of ibuprofen
Mass spectrometry techniques offer comparative advantages for speed and productivity of pharmaceutical analysis [3]. EI-MS of IB at 70 eV were recorded and investigated. A typical mass spectrum (bar graph) is shown in Fig. 2. The main fragmentation pathways after EI are presented in Scheme 1.

Ibuprofen EI-mass spectra at 70 eV

Mass proposed fragmentation route ways for formation of important fragment ions of ibuprofen drug
The spectrum is characterized by many competitive and consecutive pathways, thus forming many intense fragment ions. The mass spectrum of IB drug is also recorded using MS-MS technique [1] as illustrated in Fig. 3. This spectrum is helpful in the interpretation of the formation of some fragment ions in the IB mass spectrum.

Chromatogram and mass spectra of ibuprofen enantiomers obtained in MS/MS mode
The signal that appears at m/z = 206 (RI = 32.8%) refers to the appearance of the molecular ion [C13H18O2]+. The moderate intensity reflects the stability of the molecular ion of IB following EI. The base peak in the spectrum that appears at m/z = 91 (RI = 100%) is mainly due to a formation of tropylium ion as a secondary process as shown in Scheme 1. Two important ions are observed in the mass spectra at m/z = 161 (RI = 84%) and at m/z = 163 (RI = 73.3%). These fragment ions may be due to the rupture of COOH and propyl radical. The second prominent ion is that observed at m/z = 119 which may be due to C3H6 + loss from fragment ion of m/z = 161 (Scheme 1, route a).
Thermal analyses
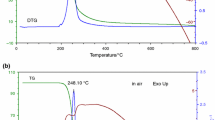
The TAs of ibuprofen are illustrated in Fig. 3. From TG/DTG (Fig. 4a), it is clear that this compound decomposed completely within the temperature range of 80–400 °C (mass loss = 99.35%). The main mass loss of this compound occurs at 191.62 °C as shown by DTG curve. From DTA curve (Fig. 4b), it is clear that thermal decomposition of ibuprofen occurs in three main endothermic regions: 80–100, 150–220, and 290–360 °C, which cannot easily be detected by TG technique. These endothermic mass losses required energy values of −214.83, −895.95, and −211.10 J g−1 at 76.06, 202.6,8 and 316.53 °C, respectively. Therefore, the proposed thermal decomposition of ibuprofen can be carried out in three consecutive steps as shown in Scheme 2.

Thermal analyses of Ibuprofen: a TG/DTG; b DTA

The proposed thermal fragmentation of ibuprofen
Computational molecular orbital (MO) calculation
The MO calculation gives valuable information about the structure and reactivity of the molecules, which actually can be used for supporting the experimental evidence. The very important parameters calculated using MO calculation includes bond orders, bond length, charge distribution, bond strain, and heat of formation. In the present study, the calculations have been carried out on ibuprofen, neutral molecule (related to TA decomposition) and charged molecular ion (related to MS fragmentation) which is used for prediction of the weakest bond rupture to follow the fragmentation pathways in both techniques.
Tables 1 and 2 represent the values of bond length/Å, bond order, and bond strain/kcal mol−1 together with dipole moment and heats of formation for ibuprofen drug molecule both in neutral and cationic forms. One can conclude the following from these tables:
-
1.
Small differences in bond length in IB system upon ionization, indicating no appreciable change in the geometries upon ionization.
-
2.
The lowest bond order (important for prediction of primary site of cleavage) observed at C8–C15 bond for both neutral (0.919) and positive species (0.901).
-
3.
Upon ionization, the stability of the molecule decreased by −206.919 kcal mol−1; which is equal to the difference between ΔH f/−101.3019 and −ΔH +f /105.617 kcal mol−1.
-
4.
From the dipole moment, electron affinity, heat of formation, and ionization potential values (Table 3) of ibuprofen in both forms of A (neutral) and B (cation); it is clear that the neutral form is the stable one with the heat of formation = −101.301 kcal mol−1; and the cation is the pharmaceutically active form, with the heat of formation = 105.617 kcal mol−1 in both in vitro and in vivo systems, i.e., it gets more easily fragmented into its metabolites.
Table 3 The dipole moment, electron affinity, heat of formation, and ionization potential values of ibuprofen in both forms A (neutral) and B (cation)
The charge distributions of different atoms (C and O) for neutral and charged species are summarized in Fig. 5. From these data, it is clear that in both IB forms, the charges on C8 and C15 are of the same sign; it means there are repulsive forces that help in causing the early rupture of this bond leading to elimination of COOH in first step in both the TA and MS techniques. These data are also used for explaining the consequent bond rupture in both techniques.

Charge distribution on different atoms of neutral (out bracket) and charged ion (in bracket)
Correlation between mass spectral fragmentation and MO calculation for charged molecule
The scope of this investigation is restricted to a search for prediction discerns features of initial bond ruptures during the course of fragmentation of compounds. Empirical observations indicate extent that the course of subsequent fragmentation is determined to large extent by the initial bond rupture of molecular ion in MS [25]. It is quite reasonable to say that the computational quantum chemistry can provide additional data information which can be used successfully for interpretation of both TA and MS results. These theoretical data can, particularly, be valuable for those MS scientists who study in gas-phase species, which can be handled much more easily by quantum chemistry than those species surrounded by solvent. Mass spectrum of IB reveals many competitive and consecutive fragmentation pathways (Scheme 1) including the principal fragmentation pathway. Representative MO calculation data are given in Table 2. From these data of the ionic form of ibuprofen cation, it is clear that the order of the bond strength is C6–C10 > C4–C8 > C22–C25 = C22–C26 > C10–C22 > C8–C13 > C8–C15. The stable bonds are C15–O19 > C15–O20. The stable bond is the one with high bond order, less bond strain, or the lowest bond length. This is the possible sequence of bond cleavage of this drug in vitro and in vivo systems.
Table 2 containing the PM3 data reveals that C8–C15 region is the first site of bond rupture; lowest bond order = 0.901, large bond length = 1.527 Å, and small bond strain = 0.037 kcal mol−1, forming fragment ion at m/z = 161, RI = 84% which is due to COOH group loss as CO2 gas (Scheme 1, route a). On the other hand, the formation of this ion (m/z = 161) from the molecular ion can also be observed through the investigation of MS-MS [1] of IB (Fig. 3). However, the weakness may be also related to electrostatic repulsion between the charge on C8 (0.082), and on C15 (0.387) which facilitates the rupture of this bond.
Correlation of the TA behavior and the MO calculation for neutral molecule
There is no study in the literature on the thermal stability of this drug with temperature changes either in vitro or in vivo systems. As indicated previously [12–15], a determination of the initial bond cleavage would be an important first step in using these calculations in a predictive manner. From the data of the MO calculations (Table 1) for IB in neutral form, it is clear that the order of the bond strength is C6–C10 > C4–C8 > C22–C25 = C22–C26 > C10–C22 > C8–C13 > C8–C15. The stable bonds are C15–O19 = C15–O20. The stable bond is the one with of high bond order; less bond strain, and the lowest bond length. This is the possible sequence of bond cleavage of this drug in vitro and in vivo systems. On the base of MO calculations, the bond order of the C8–C15 system (Table 1) refers to the possible starting decomposition of the neutral compound at C8–C15 (the lowest bond order = 0.919, large bond length 1.521 Å, or bond strain/0.031 kcal mol−1) followed by rupture of N8–C9 bond (bond order = 0.905). On the other hand, an electrostatic repulsion between charge on C8 (0.012) and charge on C15 (0.381) facilitates the rupture of this bond. This means that the carbon monoxide is the first volatile gas molecule evolved on heating IB molecule starting at 80 °C, and the energy required for this step = −21483 J g−1. This step may be followed by loss of CH2=CH–CH3 (wt. loss % = 20.4, energy = −895.95 J g−1) followed by CH2=CH (wt. loss 13.6%) which required energy = −212.10 J g−1).
Conclusions
This study provides further insights into applicability of experimental TA and MS techniques and theoretical investigation using PM3 procedure on IB drug. From the application of both practical and theoretical techniques in commitment, it is concluded that the primary fragmentation of IB by applying both experimental techniques (TA and MS) can be explained by the COOH molecular loss from the initial IB molecule. Subsequent fragmentation in MS in its fast successive processes can be rationalized and confirmed using MO-calculations. Ibuprofen can be completely dissociated in the temperature range of 80–400 °C, while the molecular ion can be fragmented after few electron volts above the IB ionization energy of 9.496 eV. The theoretical MO calculation data help in the selection of the most probable fragmentation pathway in both the TA and MS techniques. Therefore, comparison between MS and TA helps in the selection of the proper pathway representing the decomposition of this drug to give its metabolites in vivo system. This comparison is also successfully confirmed by MO calculations.
References
Cretu G, Ionicã M, Dänet AF, Aboul-Enein H, Macovei R, Buleandrä M. Separation of the enantiomers of ibuprofen by a gas chromatographic-mass spectrometric method. Acta Chromatogr. 2005;15:315–9.
Martindala W. In: Reynolds J, editors. The extra pharmacopoeia. 29th ed. London: Pharmaceutical Press; 1989. p. 743–6.
Larsen BS, McEwen CN, editors. Mass spectrometry of biological materials. New York: Marcel Dekker; 1998.
Kerns EH, Rourich RA, Volk KJ, Lee MS. Buspirone metabolite structure profile using a standard liquid chromatographic-mass spectrometric protocol. J. Chromatogr B. 1997;698(1–2):133–45.
Zayed MA, Mohamed GG, Fahmey MA. Thermal and mass spectral characterization of novel azo dyes of p-acetoamidophenol in comparison with Hammett substituent effects and molecular orbital calculation. J Therm Anal Calorim. 2011. doi:10.1007/s10973-011-1515-8.
Levsen K. Fundamental aspects of organic mass spectrometry. Weinheim: Verlag Chemie; 1978.
Bourcier S, Hoppilliard Y. Fragmentation mechanisms of protonated benzylamines. Electrospray ionisation-tandem mass spectrometry study and ab initio molecular orbital calculations. Eur J Mass Spectrom. 2003;9:351–60.
El-Gamel NEA, Hawash MF, Fahmey MA. Structure characterization and spectroscopic investigation of ciprofloxacin drug. J Therm Anal Calorim. 2011. doi:10.1007/s10973-011-1584-8.
Fahmey MA, Zayed MA. Phenolic-iodine redox products. Mass spectrometry, thermal analysis and other physicochemical methods of analyses. J Therm Anal Calorim. 2002;67:163–75.
Fahmey MA, Zayed MA, El-Shobaky HG. Study of some phenolic-iodine redox polymeric products by thermal analyses and mass spectrometry. J Therm Anal Calorim. 2005;82:137–42.
Somogyi A, Gomory A, Vekey K, Tamas J. Use of bond orders and valences for the description and prediction of primary fragmentation processes. Org Mass Spectrom. 1991;26:936–8.
Zayed MA, Fahmey MA, Hawash MF, Abdallah SAM. Structural investigation of dextromethorphane using mass spectrometry and thermal analyses combined with MO calculations. Serb J Exp Clin Res. 2011;12(1):21–8.
Zayed MA, Fahmey MA, Hawash MF. Investigation of diazepam drug using thermal analyses, mass spectra and semi-empirical MO calculations. Spectrochim Acta A. 2005;61:799–805.
Zayed MA, Hawash MF, Fahmey MA. Structure investigation of codeine drug using mass spectra, thermal analyses and semi-empirical MO calculations. Spectrochim Acta A. 2006;64:363–71.
Zayed MA, Nour El-Dien FA, Hawash MF, Fahmey MA. Mass spectra of gliclazide drug at various ion sources temperature. Its thermal behavior and molecular orbital calculations. J Therm Anal Calorim. 2010;102:305–12.
Felix FS, Cides da Silva LC, Angnes L, Matos JR. Thermal behavior study and decomposition kinetics of salbutamol under isothermal and non-isothermal conditions. J Therm Anal Calorim. 2009;95(3):877–80.
Bonato PS, Perpetua M, Del Lama FM, de Cavalho R. Enantioselective determination of ibuprofen in plasma by high-performance liquid chromatography-electrospray mass spectrometry. J Chromatogr B. 2003;796(2):413–20.
Johannsen M. Separation of enantiomers of ibuprofen on chiral stationary phases by packed column supercritical fluid chromatography. J Chromatogr A. 2001;937:135–8.
Stubberud K, Callmer K, Weslerland D. Partial filling-micellar electrokinetic chromatography optimization studies of ibuprofen, codeine and degradation products, and coupling to mass spectrometry: part II. Electrophoresis. 2003;24:1008–15.
Jabor VAP, Lanchote VL, Bonato PS. Enantioselective analysis of ibuprofen in human plasma by anionic cyclodextrin-modified electrokinetic chromatography. Electrophoresis. 2002;23:3041–7.
Agalonovic-Kustrin S, Beresford R, Razzak M. Determination of enantiomeric composition of ibuprofen in solid state mixtures of the two by DRIFT spectroscopy. Anal Chim Acta. 2000;417(1):31–9.
Stewart JJP. Optimization of parameters for semi-empirical methods I. Method. J Comput Chem. 1989;10:209–20.
Baker J. An algorithm for the location of transition states. J Comput Chem. 1986;7:385–95.
Stewart JJP. Software package MOPAC 2000. Tokyo: Fujitsu Limited; 1999.
Gergely A. Quantum chemical interpretation of a typical fragmentation in the mass spectra of benzoquinolizine derivatives. Int J Mass Spectrom Ion Phys. 1983;46:247–50.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zayed, M.A., Hawash, M.F., Fahmey, M.A. et al. Investigation of ibuprofen drug using mass spectrometry, thermal analyses, and semi-empirical molecular orbital calculation. J Therm Anal Calorim 108, 315–322 (2012). https://doi.org/10.1007/s10973-011-1876-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-011-1876-z