
Abstract
The definition as well as prediction of rock thermal behavior seems to be a quite difficult problem significantly effected by rock composition and structure. Temperature increase causes various changes of rock material (such as decomposition, oxidation, phase and polymorphic transformation, etc.). These changes are connected to thermal expansion with following appearance of tensions and cracks in minerals and rock structure. After consequential temperature decrease, developed tensions and cracks still influence the process. This study presents the application of thermogravimetric analysis, differential thermal analysis and thermomechanical analysis in characterization of selected marble thermal behavior. The texture and morphological orientation of calcite grains for marble samples was determined by optical microscopy. FTIR spectroscopy application along with X-ray diffraction (XRD) extended data about mineralogical composition. According to optical microscopy, the calcite grains show marked morphological anisotropy in one direction for some samples. Therefore, the thermal expansion had to be measured in three different (perpendicular to each other) directions. It is evident, that the effect of temperature on the final marble properties depends not only on mineralogical composition, but also on structure, texture and morphological orientation of grains. All these facts significantly influence the interpretation of differences in various marble thermal behavior.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Marbles are used principally for buildings and monuments, interior decoration, statuary, and tabletops. Marbles are the quasi-monomineral metamorphic rocks that are made up of an aggregate of calcite crystal grains. Metamorphic rocks have been modified by heat, pressure, and chemical process; exposure to these extreme conditions has altered the mineralogy, texture, and chemical composition of the rocks. There are two basic types of metamorphic rocks—foliated metamorphic rocks and non-foliated metamorphic rocks. Marble is a non-foliated metamorphic rock, but the preferred orientation of calcite in marble is often expressively developed. First of all, twinning according (0112) and translational gliding according (1011) progress during the plastic deformation of marble.
The structural and mechanical properties of marble under various conditions of pressure, temperature, and chemical solution were studied by Lepper [1], Griggs et al. [2], Turner et al. [3], Handin et al. [4], and Royer-Carfagni [5]. Yule marble is characterised by a certain systematic arrangement of the ellipsoidal grains of calcite. It has been found that the strength of the marble perpendicular to the planes of “grains” is nearly 1.6-fold greater than the strength determined parallel to the planes. Stiffness parallel to the preferential planes is greater; the relation between the corresponding values of the modulus of elasticity (E///E⊥) was approx. 1.5. Various combinations of marble parameters like mineral composition, grain size, size distribution, grain boundary geometries, or crystallographic preferred orientation lead to different properties of the marble types [6–8].
Concerning marble chemical/mineral composition, marbles usually consist of carbonates and some additional traces of ferritic oxides, quartz, clays, micas, etc. For determination of phase composition, methods of FTIR spectroscopy and X-ray powder diffraction are usually used.
For determination of the marble thermal behavior, thermal analysis is a useful tool. The application of thermal methods for study of minerals and rocks has a history as long as that of thermal analysis itself. Thermal analysis is widely used in the investigation of minerals and rocks. Application of differential thermal analysis shows that the principal changes in marble thermal decomposition occur in several temperature intervals, depending on marble composition. If there is a presence of some admixtures like additional organics or ferritic minerals at sample, the oxidation process occurs at the range of temperatures 350–700 °C. In the case of clay or quartz, endo reactions may appear at the same temperature interval. Then at temperature interval 750–1,000 °C, dissociation of carbonate usually comes up. The endothermic peak of calcite decomposition is quite intensive; so the small amount of impurities at sample can have fleet endo or exo effects; so it is sometimes impossible to detect them on DTA curve [9–12]. Also application of thermomechanical analysis is useful for characterization of thermal behavior of rocks, in particular marbles. Important parameter obtained by thermomechanical analysis is the coefficient of thermal expansion [13].
The aim of this study is to compare the thermal behavior of Czech selected marbles to well-known Italian marble Carrara, which is widely used because of its purity, isotropic properties, and large amount of literature data about its behavior under various experimental conditions. The mineralogical composition has been characterized by X-ray diffraction and FTIR spectroscopy along with optical microscopy. The thermal behavior has been characterized by simultaneous thermogravimetry and differential thermal analysis and thermomechanical analysis. Information obtained by methods used in this study should contribute to a better understanding of changes induced by heating of marbles and relationship between their structure and thermal expansion.
Samples
Four Czech marble samples were chosen for characterization of their thermal behavior. Sample 5334 is white marble from quarry Horni Lipova, sample 5335 and 10558 are gray marbles from quarry Horni Lipova (with different grain size), and sample 3714 is marble from quarry Cerna Skala u Potstejna. For a very long time, the marbles from Carrara area in the northen Alpi Apuane in Italy are used as building stones and many research workers are interested in their properties [14–17]. So this classical Italian marble from Carrara (sample 5333) was chosen for comparison with Czech selected marbles mentioned above.
Methods
Optical microscopy
Thin sections were prepared from all marble samples. The thin sections were oriented in the direction A, B, C of specimen. The orientation was arranged in three directions perpendicular to each other (coordinate system ABC). Thin sections prepared in the direction A are perpendicular to the foliation. These thin sections were observed under BX 50 OLYMPUS polarization microscope with a DVC camera connected to a PC. The photomicrographs were taken by microscope NICON ECLIPSE 80i. The analysis was performed in polarised light at 20 times magnification. Size and preferred morphological orientation of calcite grains were determined for every thin section (coordinate system ABC).
X-ray diffraction and FTIR spectroscopy
X-ray diffraction (XRD) was performed by using Bruker D8 Advance X-ray powder diffractometer. XRD patterns were obtained using position sensitive Vantec detector, Co K α radiation and working conditions 35 kV and 25 mA, 0.03° 2θ increment and time at step 1 s.
Infrared spectra were recorded on Nicolet Avatar 320 FTIR spectrometer ThermoNicolet equipped with a DTGS/KBr detector. The KBr pressed-disk technique (2 mg of sample and 200 mg of KBr) was used. The spectra were measured by accumulating 64 scans at 4 cm−1 resolution in the spectral range from 4,000 to 400 cm−1.
Simultaneous thermogravimetry and differential thermal analysis
Thermal properties were investigated by simultaneous thermogravimetry and differential thermal analysis using a thermal analyzer Setsys Setaram with thermal measurement head TG ATD Rod. The TG/DTA curves were recorded in a static air atmosphere at a heating rate 10 and 30 °C min−1 to the final temperature 1,000 °C with sample mass 50 mg.
The kinetics of decomposition was studied using the Arrhenius equation applied to solid state reactions. The activation energy for decomposition of CaCO3 was evaluated by Freeman–Carroll method [18] which enables, from the exploitation of one DTG peak, to determine besides activation energy of reaction also the reaction order and the rate constant. The possibilities and comparison of Freeman–Carroll method with other used methods are described well by Slovak [19].
Thermomechanical analysis
Thermal expansion was determined by thermomechanical analysis using the thermal analyzer Setsys Setaram with thermal measurement head TMA quartz. Test specimens (cube 10 × 10 × 10 mm) were heated in a furnace, and the displacement was transmitted by means of QUARTZ probe. The probe was a spherical-ended with a 5 mm diameter. Measuring of deformation was performed by compression with a probe resting on the sample and the load adjusted up to nil value. Sample cubes were prepared according to the three principal axes of the reference coordinate system ABC mentioned above. Resultant TMA curves show the measurement of a specimen’s dimension (length) as a function of temperature under constant compressive load and following conditions: final temperature 800 °C, heating rate 30 °C min−1. This technique was used to determine residual thermal expansion (ε), thermal expansion obtained during heating (εH), and the coefficient of thermal expansion (αH) in the temperature interval 25–800 °C.
Results and discussion
Optical microscopy
Optical microscopy was used to evaluate the average size of calcite grains in marble samples as well as the priority morphological orientation (PMO) of calcite grains. This method was also used to confirm the composition of analyzed samples. The average size of calcite grains in marble samples is summarized in Table 1 along with PMO. The intensity of PMO was divided into 4 stages: 0 as invisible, + as partial, ++ as visible, and +++ as expressive. In case of mineralogy composition, the calcite was found as the main phase for all analyzed samples. Pyrite was found as the admixture for two gray marble samples 5335 and 10558. Sample 5335 showed pyrite even in crystal modification, and its amount was significantly higher than for sample 10558 (Fig. 1b). For illustration, the photomicrographs of marble microstructure of samples 5335 and 5333 are shown in Figs. 1 and 2.

a Photomicrographs of the marble microstructure—thin section no. 5335 A. b Pyrite crystal (transmitted light, crossed polarizers)

Photomicrographs of the marble microstructure—thin section no. 5333 A (transmitted light, crossed polarizers)
XRD and FTIR measurements
X-ray diffraction analysis was used to evaluate patterns considering the phase analysis and profile analysis. The phase analysis did not reveal any evident differences in quality of the sample. Major phase was calcite for all analyzed samples with next constituents like quartz, feldspar, muscovite in a very small quantity.
However, the profile analysis, observing intensity, and relation of interlayer space d104 and broadening of this diffraction, the differentiation of the samples were clear (Fig. 3). The samples 5335 and 10588 are very similar in this case, while sample 3714 is remarkably different. Diffraction peak of the sample 3714 is the lowest intensity and develops two unclear peaks’ maxima; therefore, the broadening of the peak is high. The samples 5334 and 5333 can be regarded as similar evaluating the position of the peak, but diffraction intensity of 5334 is higher than that of 5335. In a typical XRD pattern (samples 5335 and 5333), the presence of calcite and small amount of SiO2 is shown (Figs. 4, 5).

XRD profile analysis of marble samples

XRD phase analysis of marble sample 5335

XRD phase analysis of marble sample 5333
IR spectra of studied samples confirmed the presence of major compound carbonate (bands at 1420, 875, 712 cm−1) with only minor quartz (bands at 1180, 1124, 1081 cm−1) and illite, muscovite (bands at 3440, 1028, 1021 cm−1) admixtures. The measured IR spectra were compared with records of published spectra [20, 21]. The results from FTIR correspond well with these from XRD. For illustrative purposes, IR spectra of marble samples (5335 and 5333) are given in Figs. 6 and 7.

FTIR spectrum of sample 5335

FTIR spectrum of sample 5333
TG and DTA measurements
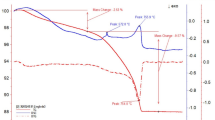
The thermal stability of marbles was studied by simultaneous thermogravimetry and differential thermal analysis. The measurements were carried out with two heating rates (10 and 30 °C min−1). Figure 8 shows the derivative thermogravimetric (DTG) curves for all used marble samples for heating rate 10 °C min−1. With increase of peak temperature on DTG curve, activation energy of respective carbonate dissociation decreased. The values of activation energy E a varied from 227 kJ mol−1 (sample 5334) to 266 kJ mol−1 (sample 5335) with the reaction order n around 0.3 for heating rate 10 °C min−1 and E a varied from 307 kJ mol−1 (sample 5334) to 375 kJ mol−1 (sample 5335) with the reaction order n around 0.7 for heating rate 30 °C min−1.

DTG curves of marble samples measured under 10 °C min−1
The shape and temperature of peaks corresponding with calcite dissociation on DTA curves are very close. DTA curves confirmed the presence of organic matter (graphite) in both gray marble samples from Lipova quarry (sample 5335 and 10558) by low intensive exothermal effect at temperature interval 650–750 °C. It is common graphite substance with quite low orderliness abounded at gray marbles from Horni Lipova quarry. Also pyrite was found as an associated mineral in sample 5335 and 10558. The exotherm peak of low intensity at about 337 °C accordant with polymorphic transformation was found as well as the exotherm peak (quite high intensity with quite low mass loss) at about 492 °C accordant with pyrite oxidation. The endotherm effect of low intensity at about 640 °C accordant with changes Fe2O3 to Fe3O4 was not found because of the graphite oxidation effect at the same temperature interval. The mass loss corresponding to both graphite oxidation and pyrite oxidation/decomposition was about 1–1.5%.
The presence of both admixtures was confirmed by optical microscopy, and the presence of pyrite was confirmed by XRD phase analysis too. The other admixtures determined by XRD and FTIR could not be confirmed by simultaneous TG/DTA because of their really small amount and overlapping of illite/muscovite crystal lattice breaks and recrystallization effects with carbonate dissociation effect. According to DTA curves, calcite was found as a dominant compound. The values of carbonate volume obtained from thermogravimetry curves measured several times under two different heating rates are shown in Table 2. The DTA peak temperatures increase with increasing of calcite volume. The lowest calcite volume was found for sample 5335, which displays the presence of pyrite and graphite substance in sample. For illustrative purposes, DTA curves of marble samples (5335 and 5333) measured at heating rate 10 °C min−1 are given in Figs. 9 and 10. The peak temperatures of carbonate dissociation obtained from DTA curves and temperatures of maximal reaction velocity obtained from DTG curves are along with the kinetics parameters (activation energy, reaction order, and correlation factor) summarized in Table 2 (comparing for heating rate 10 and 30 °C min−1). As expected, the values of peak temperatures and activation energy obtained by heating rate 10 °C min−1 were smaller, but the trend remained the same.

TG/DTA curves of sample 5335

TG/DTA curves of sample 5333
TMA measurements
Test specimens (cube 10 × 10 × 10 mm) were heated up to 800 °C and cooled down to the room temperature. The heating rate 30 °C min−1 was chosen because of elimination of disintegrating of marble cubes under lower heating rates [8, 22]. Low heating rates can bring the complications with resolving thermomechanical curves. Each marble sample was measured in three perpendicular directions A, B, C because of an anisotropy of calcite physical properties. The temperature, time, and deformation were recorded during heating/cooling. This allowed to calculate the relative dilatation ε (%) as a temperature normalized value. Also the changes in the length of the sample during heating (εH) were calculated. The coefficient of thermal expansion of the specimen was obtained by the following relationship : α.l 0 = dl/dT (where α. is the coefficient of thermal expansion, l 0 is the original sample length, and dl/dT is the rate of change of sample length with temperature). Calculated values of coefficient of linear thermal expansion αH in temperature range from 25 to 800 °C corresponded to values of thermal expansion (εH) obtained from TMA curves at the same temperature interval. Percentage displacements for tested samples are shown in Table 3.
Our results demonstrate that the strongest deformation was generally found in direction A for all tested samples, the weakest deformation was found in direction C (Figs. 11, 12, 13, 14, 15). The more fine-grained samples showed the higher value of residual thermal expansion ε than more coarse-grained samples in direction A. The sample 5334 which displayed the largest grain size value had the smallest value of residual dilatation ε, samples 5335 and 5333 with the smallest grain size values displayed the strongest residual dilatation in direction A. Sample 3714 could be clearly classified as the sample with the maximal anisotropy (the strongest difference between thermal expansion measured during heating (εH) in its ABC coordinate system), contrariwise the sample 5333 was found as sample with minimal difference between εH measured in its ABC coordinate system. For sample 3714, the strongest dilatation εH (3.36%) was observed in direction A and the weakest dilatation εH (0.47%) was observed in direction C. For sample 5333, the strongest dilatation εH (2.42%) was observed in direction A and the weakest dilatation εH (2.13%) was observed in direction C.

TMA curve of sample 5334

TMA curve of sample 5335

TMA curve of sample 10558

TMA curve of sample 3714

TMA curve of sample 5333
The results from calculation of relative dilatation ε (%) and dilatation of heating εH (%) do not show any dependance. In the case of the relative dilatation ε (%), the values increase with the decrease of calcite volume and the grain size. With regard to the values of dilatation of heating εH, the data correspond well with the results obtained by optical microscopy. Samples with the highest values of εH as well as with the coefficient of linear thermal expansion αH display also the greatest preferred orientation of calcite grains.
Conclusions
From simultaneous thermogravimetry and differential thermal analysis follows that the marble composition and thermal behavior are quite similar for all used samples. The shape and temperature of peaks corresponding with calcite dissociation on DTA curves are very close. The values of DTA peak temperatures increase with increasing of calcite volume. With the increase of peak temperature on DTG curves, the activation energy of respective carbonate dissociation decreases.
On the basis of the values of linear thermal expansion coefficient αH and the values of both dilatation ε and εH, the anisotropy of some marble samples is evident. The residual thermal dilatation clearly depends on the direction. The thermal expansion in the direction A is evidently higher than thermal expansion in directions B and C for all measured samples. The marble Carrara is generally considered to have quite isotropic physical properties (our results confirmed this fact), and the sample 5334 (white marble from Horni Lipova quarry) shows also quite isotropic physical properties, although the grain size is greater than for Carrara sample. Both samples display the lowest differences between thermal expansion in their ABC coordinate system, and both display very similar thermal behavior. Gray marble samples 5335 and 10558 from Horni Lipova quarry have very close composition including graphite and pyrite and display also the same thermal behavior with middle difference between thermal expansion in their ABC coordinate system. The strongest difference between thermal expansion in its ABC coordinate system is found for sample 3714 (marble from Cerna Skala quarry). This sample also shows the strongest dilatation εH as well as the highest coefficient of linear expansion αH in direction A from all tested samples.
The marked anisotropy of thermal expansion is evident for samples with visible or extensive PMO of calcite grains. The low or no anisotropy of thermal expansion is evident for samples with partial or invisible priority. In the case of different values of thermal expansion in all three measured directions, the strongest values are found in the direction normal to foliation (direction A) which also corresponds with the literature data [6]. According to the evaluated optical and thermal properties it is possible to divide the marble samples into three basic groups with different anisotropy: low (samples 5334 and 5333), middle (samples 5335 and 10558), and strong (sample 3714).
References
Lepper HA Jr. Compression tests on oriented specimens of Yule marble. Am J Sci. 1949;247:570–75 (in Kwaśniewski 1993).
Griggs D, Miller WB. Deformation of Yule marble: part I. Geol Soc Am Bull 1951;62:853–62 (in Kwaśniewski 1993).
Turner FJ, Griggs D, Heard HC. Experimental deformation of calcite crystals. Geol Soc Am Bull. 1954;65:883–934.
Handin J, Higgs D, O’Brien JK. Torsion of Yule marble under confining pressure. Geol Soc Am Meml. 1960;79:245–74.
Royer-Carfagni GF. On the thermal degradation of marble. Rock Mech Min Sci. 1998;36:119–26.
Leiss B, Weiss T. Fabric anisotropy and its influence on physical weathering of different types of Carrara marbles. J Struct Geol. 2000;22:1737–45.
Siegesmund S, et al. Physical weathering of marbles caused by anisotropic thermal expansion. Int J Earth Sci. 2000;89:170–82.
Obara B, Kožušníková A. Utilisation of the image analysis method for the detection of the morphological anisotropy of calcite grains in marble. Comput Geosci. 2007;11(4):275–81.
Dagounaki C, et al. Thermal characterization of carbonate rocks, Kozani area, north-western Macedonia, Greece. J Therm Anal Calorim. 2004;78:295–306.
Gokhale KV, Rao TC. Studies on thermal dissociation of some carbonate minerals. J Therm Anal. 1970;2:83–5.
Hatakeyama T, Liu Z. Handbook of thermal analysis. New York: Wiley; 1998.
Sanders JP, Gallagher PK. Kinetic analyses simultaneous TG/DSC measurements. J Therm Anal Calorim. 2005;82:659–64.
Price DM. Principles of thermal analysis and calorimetry. Cambridge: The Royal Society of Chemistry; 1994.
Barsotelli M, Fratini F, Giorgetti G. Microfabric and alteration in Carrara marble: a preliminary study. Sci Technol Cult Herit. 1999;7:115–26.
Pierri M, et al. Rheological and microstructural evolution of Carrara marble with high shear strain: results from high temperature torsion experiments. J Struct Geol. 2001;23:1393–413.
Leiss B, Molli G. High-temperature texture in naturally deformed Carrara marble from Alpi Apuane, Italy. J Struct Geol. 2003;25:649–58.
Oesterling N, et al. Strain dependent variation of microstructure and texture in naturally deformed Carrara marble. J Struct Geol. 2007;29:681–96.
Freeman ES, Caroll B. The application of thermoanalytical techniques to reaction kinetics: the thermogravimetric evaluation of the kinetics of the decomposition of calcium oxalate monohydrate. J Phys Chem. 1958;62:394–9.
Slovak V. Determination of kinetics parameters by direct non-linear regression from TG curves. Thermochim Acta. 2001;372:175–82.
Farmer VC. The infrared spectra of minerals. London: Mineralogical Society; 1974.
Madejova J, Komadel P. Baseline studies of the clay minerals society source clays: infrared methods. Clay Clays Miner. 2001;49(5):410–32.
Barnhoorn A, et al. The role of recrystallisation on the deformation of calcite rock. J Struct Geol. 2004;26:885–903.
Acknowledgements
This study was supported by the Czech Science Foundation, the project No.105/07/P416, the project No. 105/08/1398 and by Research plan No. AVOZ 30860518. The authors would like to thank George Laynr for controlling and correcting the use of English in this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Plevová, E., Kožušníková, A., Vaculíková, L. et al. Thermal behavior of selected Czech marble samples. J Therm Anal Calorim 101, 657–664 (2010). https://doi.org/10.1007/s10973-010-0907-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-010-0907-5