
Abstract
The thermal decomposition of magnesium hydrogen phosphate trihydrate MgHPO4 · 3H2O was investigated in air atmosphere using TG-DTG-DTA. MgHPO4 · 3H2O decomposes in a single step and its final decomposition product (Mg2P2O7) was obtained. The activation energies of the decomposition step of MgHPO4 · 3H2O were calculated through the isoconversional methods of the Ozawa, Kissinger–Akahira–Sunose (KAS) and Iterative equation, and the possible conversion function has been estimated through the Coats and Redfern integral equation. The activation energies calculated for the decomposition reaction by different techniques and methods were found to be consistent. The better kinetic model of the decomposition reaction for MgHPO4 · 3H2O is the F 1/3 model as a simple n-order reaction of “chemical process or mechanism no-invoking equation”. The thermodynamic functions (ΔH*, ΔG* and ΔS*) of the decomposition reaction are calculated by the activated complex theory and indicate that the process is non-spontaneous without connecting with the introduction of heat.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnesium phosphate compounds are widely used in several fields of industry, for example in agricultural, pharmaceutical, food, textile industry, etc. [1]. Thermal treatments of many metal phosphates have a great synthetic potential as it may turn simple compounds into advanced materials, which relate to the hydrolysis and dehydration reactions at elevated temperatures [2]. Studies on thermodynamics, mechanisms and kinetics of solid-state reactions are challenging and difficult task with complexity resulting from the great variety of factors with diverse effects, e.g., reconstruction of solid state crystal lattice, formation and growth of new crystallization nuclei, diffusion of gaseous reagents or reaction products, materials heat conductance, static or dynamic character of the environment, physical state of the reagents—dispersity, layer thickness, specific area and porosity, type, amount and distribution of the active centers on solid state surface, etc. [3–5]. Recently, the thermal analysis (TA) methods have been widely used for scientific and practical purposes due to they provide reliable information on the physico-chemical parameters characterizing the processes of isothermal or non-isothermal decompositions [6]. The results obtained on these bases can be directly applied in materials science for the preparation of various metals and alloys, cements, ceramics, glasses, enamels, glazes, polymer and composite materials.
In this respect, MgHPO4 · 3H2O and Mg2P2O7 are among the most widespread of mineral phosphates and the synthetic compounds [1, 2]. These magnesium phosphates are also used as anticorrosive inorganic pigments because their main properties are insolubility in water, high temperature resistance and chemical stability [2]. The advantage of these magnesium phosphates are the absence of ecologically (toxicologically) harmful elements. Thus, in the last few years many works have undertaken a series of research studies on the synthesis, characterization and application activities of MgHPO4 · 3H2O and Mg2P2O7 [1]. However, the thermodynamic functions, mechanism and kinetic parameters of thermal decomposition of MgHPO4 · 3H2O for the synthesize Mg2P2O7 have not been reported elsewhere. This is especially important for establishing the interrelation between the structure of vitreous (and crystalline) pyrophosphates formed in this system, its composition, and different physico-chemical properties [3–5].
The aim of the present paper is to study the kinetic and thermodynamic parameters of the formation of Mg2P2O7 from the thermal transformation of MgHPO4 · 3H2O and is followed using differential thermal analysis-thermogravimetry (TG-DTG-DTA). Non-isothermal kinetic of the decomposition process of MgHPO4 · 3H2O was interpreted by the Ozawa [7] and Kissinger-Akahira-Sunose [8, 9] and iterative methods [10, reference therein]. The possible conversion function has been estimated using the Coats and Redfern method which gives the best description of the studied decomposition process and allows the calculation of reliable values of kinetic triplet parameter [11]. The thermodynamic (∆H*, ∆S*, ∆G*) and kinetic (E, A, mechanism and model) parameters of the decomposition reaction of MgHPO4 · 3H2O have attracted the interesting of thermodynamic and kinetic scientists, which are discussed for the first time.
Experimental
Materials and measurement
MgHPO4 · 3H2O crystalline powder (34,075-8; 99%) was commercially obtained from Aldrich Co. Ltd and was used without further purification. The characterization and identification of MgHPO4 · 3H2O and its final decomposition product Mg2P2O7 were carried out by X-ray powder diffraction [2] (a Siemens D500 diffractometer) and scanning electron microscope [2] (a JEOL JSM-5400). Thermal analysis measurements (thermogravimetry, TG; differential thermogravimetry, DTG; and differential thermal analysis, DTA) were carried out on a Pyris Diamond Perkin Elmer apparatus by increasing temperature from 303 to 1,273 K with calcined α-Al2O3 powder as the standard reference. The experiments were performed in air atmosphere at heating rates of 5, 10, 15, and 20 K min−1. The sample mass was kept about 6.0–10.0 mg in an alumina crucible without pressing. The room temperature FTIR spectra were recorded in the range of 4,000–370 cm−1 with eight scans on a Perkin-Elmer Spectrum GX FT-IR/FT-Raman spectrometer with the resolution of 4 cm−1 using KBr pellets (spectroscopy grade, Merck).
Kinetics studies
Thermal transformation of crystal hydrates is a solid-state process of the type [12–18]: A (solid) → B (solid) + C (gas). The kinetics of such reactions is described by various equations taking into account the special features of their mechanisms. The reaction can be expressed through the temperatures corresponding to fixed values of the extent of conversion (α = (m i − m t)/(m t − m f), where m i, m t and m f are the initial, current and final sample mass, at moment time t) from experiments at different heating rates (β).
The temperature dependence of the rate constant k for the process is described by the Arrhenius equation:
where A is the pre-exponential factor (s−1), E is the apparent activation energy (kJ mol−1), T is the absolute temperature (K) and R is the gas constant (8.314 kJ mol−1 K−1). Substitution of Eq. 2 in Eq. 1 gives:
When the temperature increases at a constant rate,
Therefore:
The conversion function f(α) for a solid-state reaction depends on the reaction mechanism. The solutions of the left hand side integral depend on the explicit expression of the function f(α) and are denoted as g(α). The formal expressions of the functions g(α) depend on the conversion mechanism and its mathematical model. The latter usually represents the limiting stage of the reaction—the chemical reactions; random nucleation and nuclei growth; phase boundary reaction or diffusion. Algebraic expressions of functions of the most common reaction mechanisms operating in solid-state reactions are presented in literatures [12–19].
Calculation of the activation energy
The activation energy (E α) can be calculated according to the isoconversional methods. In kinetic study of MgHPO4 · 3H2O, the Ozawa [7] and Kissinger–Akahira–Sunose (KAS) [8, 9] equations were used to determine the activation energy of the dehydration reaction.
The equations used for E α calculation are:
Ozawa equation:
KAS equation:
The Arrhenius parameters, together with the reaction model, are sometimes called the kinetic triplet. \( g(\alpha ) = \int\limits_{0}^{\alpha } {{\frac{{{\text{d}}\alpha }}{f(\alpha )}}} \) is the integral form of the reciprocal of f(α), which is the reaction model and depends on the reaction mechanism. According to these equations, the reaction mechanism and the shape of g(α) function cannot affect the calculation of the activation energies of the different stages. Iterative procedure is used to calculate the approximate value of E approach to the exact value, according to the equations are [10]
where h(x) is expressed by the fourth Senum and Yang approximation formulae:
where x = E α/RT, E α is the average activation energy from KAS method
The iterative procedure is as follows: (i) suppose h(x) = 1 to estimate the initial value of the activation energy E α1. The conventional isoconversional methods stop the calculation at this step; (ii) using E α1 to calculate h(x), then from Eq. 8 and 9 to calculate a new value of E α2 for the activation energy from the plot of ln[β/h(x)T 2] versus 1/T; (iii) repeat step (ii), replacing E α1 with E α2. When E α1 − E α2 < 0.1 kJ mol−1, the last value of E α1 is the exact value of the activation energy of the reaction. The plot is model independent since the estimation of the apparent activation energy does not require selection of particular kinetic model (type of g(α) function), which indicates that the activation energy values are usually regarded as more reliable than these obtained by a single TG curve.
Estimation of the conversion function and the pre-exponential factor
Several authors [6–14] suggested different ways to solve the right hand side integral in Eq. 5. For the present study, one calculation procedure was based on the Coats and Redfern equation [11]. Data from TG curves (Fig. 1 and Table 1) in the decomposition range 0.2 < α < 0.8 were used to determine the kinetic parameters of the process in all used calculation procedures. The integral method of the Coats and Redfern has been mostly and successfully used for studying of the kinetics of dehydration and decomposition of different solid substances [17–19]. The kinetic parameters can be derived using a linear form of the modified Coats and Redfern equation:

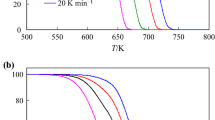
TG-DTG-DTA curves of MgHPO4 · 3H2O in air at four heating rates (5, 10, 15 and 20 K min−1)
Hence, \( \ln \left( {{\frac{g(\alpha )}{{T^{2} }}}} \right) \) calculated for the different α values at the single β value on 1000/T must give rise to a single master straight line, so the activation energy and the pre-exponential factor can be calculated from the slope and intercept through ordinary least square estimation. The activation energy, pre-exponential factor and the correlation coefficient can be calculated from the equation of the Coats and Redfern combined with 23 conversion functions [14–18]. Comparing the kinetic parameters from the Coats and Redfern equation, the probable kinetic model may be selected, which the values of E α and A were calculated with the better linear correlation coefficient and the activation energies obtained from the Coats and Redfern equation above were shown in good agreement to those obtained from iterative procedure or KAS or Ozawa methods with a better correlation coefficient (r 2). So the pre-exponential factor A can be obtained by calculating the average value of A(s−1) from Eq. 10 for different heating rates.
Estimation of the thermodynamic functions
From the activated complex theory (transition state) of Eyring [12, 13, 19–21], the following general equation may be written:
where A is the pre-exponential factor A obtained from the Coats and Redfern method; e = 2.7183 is the Neper number; χ: transition factor, which is unity for monomolecular reactions; k B: Boltzmann constant; h: Plank constant, and T p is the peak temperature of DTA curve. The change of the entropy may be calculated according to the formula:
Since
when E* is the activation energy E a obtained from the iterative method. The changes of the enthalpy ΔH* and Gibbs free energy ΔG* for the activated complex formation from the reagent can be calculated using the well-known thermodynamic equation:
The heat of activation (∆H*), entropy of activation (∆S*), free energy of activation decomposition (∆G*) were calculated at T = T p (T p is the DTA peak temperature at the corresponding stage), since this temperature characterizes the highest rate of the process, and therefore, is its important parameter.
Results and discussion
Thermal analysis
TG-DTG-DTA curves of the thermal decomposition of MgHPO4 · 3H2O at four heating rates are shown in Fig. 1. All curves are approximately the same shape and indicate the mass loss is independent of the heating rate. All TG curves show that a single step of the mass loss in the range of 400–623 K. The mass loss in this range depends on the heating rate: the mass loss increases with the decreasing of the heating rate. The increasing mass loss at the beginning of all TG curves is observed, which may be assumed that the studied compound adsorbed moisture. The mass loss at all four heating rates is between 32.74 and 36.50% (3.17–3.53 mol H2O), which agrees with to the theoretical value for MgHPO4 · 3H2O (36.14%, 3.50 H2O). A single peak in DTA and DTG curves (442 K) closely correspond to the mass loss observed in TG traces. Additionally, the decomposition stage was shifted toward higher temperature when the heating rates increase. The mass retained is about 64.00% for all heating rates, compatible with the value expected for the formation of Mg2P2O7, which is verified by XRD [2] and FTIR (Fig. 2) measurements. The overall reaction is:
Subsequently, it has been shown that the water of crystallization cannot be usually removed without the destruction of anion structure. Therefore, the dehydration of crystallization water (3.0 mol H2O) and an intramolecular dehydration of the protonated hydrogen phosphate group (0.5 mol H2O) are often overlapped according to the scheme given (15), which is consistent with a single decomposed step (Fig. 1). The water in crystalline hydrate may be considered either as crystal water or as co-ordinated water. The strength of the binding of these molecules in the crystal lattice is different, hence, resulting in different dehydration temperatures. The temperature at which theoretical mass loss is achieved and can be also determined from the TG curve and considered to be the minimum temperature needed for the calcinations process. Thus, MgHPO4 · 3H2O sample was calcined at 673 K for 2 h in the furnace.

FTIR spectra of MgHPO4 · 3H2O (a) and its dehydrated product Mg2P2O7 (b)
The specificity of the thermal decomposition was characterized by identification of the bonds to be selectively activated due to energy absorption at vibrational level [14]. These bonds were assigned by comparing the calculated wavenumbers with the observed wavenumbers in the IR spectra. This breaking bond is assimilated with a Morse oscillators [22] coupled non-linear [23] with the harmonic oscillators of the thermic field. Following, a theoretical treatment developed by Vlase et al. [14], this work apply the relation between the average maximum temperature peak T p (DTA) at four heating rates (Fig. 1) and the wavenumber of activated bond is calculated by:
where \( k_{\text{b}} \,{\text{and}}\,h \) are, respectively, the Boltzmann and Planck constants, and c the light velocity. Because the breaking bond has an unharmonic behavior, the specific activation is possible due to more than one quanta, or by a higher harmonic. Here, ωcal is calucated by Eq. 16, which is assigned the spectroscopic number for the bond supposed to break. ωsp is the frequency bands of vibrational modes and is calculated by \( \omega_{\text{sp}} = q\omega_{\text{calc}} ,\,q \in N\, = \,1,2,3, \ldots ,\left( {\text{quanta number}} \right) \). Additionally, the ωcal values with the ωsp values determined from the DTA curves at four heating rates (5, 10, 15 and 20 K min−1), together with the assignments of the corresponding oscillations are compared. In order to corroborate the calculated data with the spectroscopic ones, we drew up the FT-IR spectra of the studied compound (Fig. 2) [2]. The average T p peak at four heating rates is 420 K. The calculated harmonic energy (ω calc) values calculated from T p peak are 614 (q = 2), 876 (3), 1168 (4), 1753 (6), 2337 (8), 2922 (10), 3214 (11) and 3506 (12) cm−1. These wavenumbers can be assigned δ(POH), ν3(PO4), C band (νOH HPO −24 ), B band (νOH HPO −24 ), A band (νOH HPO −24 ) and νOH (ν1 and ν3 H2O) vibrations, respectively [23, 24]. These results support that the overlapped of the elimination of water of crystallization and the destruction of HPO4 2− anion structure [2, 24], which confirm single decomposition step in Eq. 15. The studied compound exhibited a very good agreement between the calculated wavenumbers from average T p (DTA) and the observed wavenumbers from IR spectra (Fig. 2) for the bonds suggested being broken. These results indicate the use of T p (DTA) will be an alternative method for the calculated wave numbers for identification in each thermal transition step of interesting materials.
FT-IR spectroscopy
The FT-IR spectra of MgHPO4 · 3H2O and Mg2P2O7 are shown in Fig. 2, which are very similar to those of MgHPO4 · 3H2O and Mg2P2O7 [2, 24]. Vibrational bands are identified in relation to the crystal structure in terms of the fundamental vibrating units namely \( {\text{HPO}}_{4}^{ - 2}\) and H2O for MgHPO4 · 3H2O and [P2O7]4− ion for Mg2P2O7, which are assigned according to the literatures [2, 24]. Vibrational bands of \( {\text{HPO}}_{4}^{ - 2} \) ion are observed in the regions of 300–500, 700–900, 1160–900, 840–930, 1000–1200, 2300–2400, 2800–3120, and 3200–3500 cm−1 These bands are assigned to the δ(PO3), γ(POH), δ(POH), ν(PO2(OH)), ν(PO3), B band \( \left( {\nu_{\text{OH}} {\text{ HPO}}_{4}^{ - 2} } \right) \), A band \( \left( {\nu_{\text{OH}} {\text{ HPO}}_{4}^{ - 2} } \right)\,{\text{and }}\nu_{\text{OH }} \left( {\nu_{{ 1 }} {\text{and}}_{ ~ } \nu_{ 3} {\text{H}}_{ 2} {\text{O}}} \right) \), respectively. The observed bands in 1,600–1,700 cm−1 region are attributed to the water bending/C band. The P–O stretching modes of the [P2O7]4− anion are known to appear in the 1,150–960 cm−1 region [2, 3, 24]. The symmetric PO2 stretching vibrations (ν sym PO2) is observed at 978 cm−1, while the asymmetric stretching vibrations (ν asym PO2) is located at 1,105 cm−1. The symmetric stretch (ν sym POP) bridge vibration for this salt is observed at 734 cm−1. The PO3 deformation and rocking modes, the POP deformations as well as the torsional and external modes are found in the 581–429 cm−1 region.
Kinetic studies
Calculation of the activation energy by isoconversional method
According to linear isoconversional method, the basic data of α and T collected from the TG curves (Fig. 1) of the decomposition step of MgHPO4 · 3H2O at various heating rates (5, 10, 15 and 20 K min−1) are illustrated in Table 1. According to the Eqs. 6–8, the plots of ln β versus 1000/T (Ozawa), ln β/T 2 versus 1000/T (KAS) and ln β/(h(x)T 2) versus 1000/T (iterative method) corresponding to different conversions α can be obtained by a linear regression of least-square method, respectively. The Ozawa, KAS and iterative analysis results of four TG measurements below 523 K are presented in Fig. 3 (a, b and c), respectively. The activation energies E α can be calculated from the slopes of the straight lines with better linear correlation coefficient (r 2) in the α range of 0.2–0.8. As shown in Table 2, the calculated activation energy values show a considerable nearly for all methods. If E α values are independent of α, the decomposition may be a simple reaction, [19–21] while the dependence of E α on α should be interpreted in terms of multi-step reaction mechanisms [14–16]. Neglecting the dependence of E versus α, an average values of E α = 100 ± 3.27 kJ mol−1 (Ozawa), 113.05 ± 2.11 kJ mol−1 (KAS) and 102.00 ± 2.36 kJ mol−1 (Iterative method) are obtained. It was considered that the E α values are independent of α if the relative error of the slope of Ozawa, KAS and iterative equations straight lines is lower than 10%. The KAS method is an exception that gives, in comparison with the other methods, considerably higher activation energy values. Then, it can be concluded that the activation energy values obtained from Ozawa or iterative methods are more reliable and these methods can be used in the determination of the activation energy values with satisfactory accuracy. For the decomposition reaction of MgHPO4 · 3H2O, the activation energy values change little with α, so this process could be a single kinetic mechanism. The activation energy for the release of the water of crystallization lie in the range of 60–80 kJ mol−1, while the values for coordinately bounded decomposition are within the range of 130–160 kJ mol−1 [14–16, 19–21]. In addition, the water eliminated at 423 K and below can be considered as water of crystallization, whereas water eliminated at 473 K and above indicates its co-ordination by the metal atom [14–16]. The calculated activation energies from the Ozawa, KAS and iterative methods for the decomposition reaction lie in the range of 100–113 kJ mol−1 whereas the decomposition stage of MgHPO4 · 3H2O is observed in the temperature range of 400–473 K. The activation energy for the decomposition reaction of MgHPO4 · 3H2O is smaller than values of MnHPO4 · H2O (~302 kJ mol−1) and some manganese phosphate hydrates (>200 kJ mol−1). Additionally, the activation energies for the decomposition reaction of MnHPO4 · H2O vary with α (0.2–0.8), which indicate a multi-step mechanism so the Coats–Redfern method for the determination of the most probably function can not be applied [24]. These differences support the cooperative effect, which relates with metal size. These activation energies are related to the vibration frequencies and are the indication of the energy of the breaking bond of intermediate species. On the basis of the activation energy values, the single decomposition mechanism of the studied compound relates the dehydration reaction of the water of crystallization and the deprotonated hydrogen phosphate (Eq. 15).

Ozawa (a), KAS (b) and iterative (c) analysis of four TG measurements for the dehydration step of MgHPO4 · 3H2O
Determination of the most probably mechanism
The following equation was used to estimate the most correct mechanism, i.e., g(α) and f(α) functions. The activation energy, pre-exponential factor and the correlation coefficient can be calculated from the Coats and Redfern equation combined with 23 conversion functions [13, 15]. The most probable mechanism function was assumed to be the one for which the value of the correlation coefficient was higher and the activation energies calculated by the Coats and Redfern method were close to the optimized values from the Ozawa, KAS and iterative methods. The optimized values from the Coats and Redfern method are the data of activation energy and pre-exponential factor, those were calculated with the best equation and are shown in Table 3. According to Table 3, it was seen that the activation energy values calculated by the Ozawa, KAS and iterative methods were close to the optimized values from the Coats and Redfern method, and the respective correlation coefficients are preferable. This is considered enough to conclude that the non-isothermal kinetic parameters of dehydration reaction of MgHPO4 · 3H2O can be reliably calculated with correctly chosen g(α) and f(α) functions. Based on these results, we can draw a conclusion that the obtained possible conversion function is F1/3 model (chemical process or mechanism no-invoking equation of one-third order) for the dehydration of MgHPO4 · 3H2O, and the corresponding function is f(α) = (3/2)(1−α)]1/3 and g(α) = 1−(1−α)]2/3. The correlated kinetic parameters are E α = 108.14 ± 11.09 kJmol−1 and A = 5.50 × 109 ± 1.0 s−1 (r 2 = 0.99107). The most kinetic function for decomposition reaction of the studied compound is different from some metal phosphate hydrates [3, 21, 24–26], which confirms the specific effect of each material. Based on the values of activation energy and pre-exponential factor, the strengths of binding of water molecules in the crystal lattice are different and, hence, results in different dehydration temperatures and kinetic parameters [19–21]. The activation energy of the decomposition reaction calculated by using F1/3 model (Table 3) that suggests the hydrates (3.5 mol H2O) in the studied compound are the different water types in the structure. This result indicates that the elimination of water obtain from 3 mol H2O for the water of crystallization and 0.5 mol H2O for the deprotonated hydrogen phosphate (HPO4 2−) and the mechanism reaction appear in the single step. The pre-exponential factor (A) values in Arrhenius equation for solid phase reactions are expected to be in a wide range (six or seven orders of magnitude), even after the effect of surface area is taken into account [13, 14]. The low factors will often indicate a surface reaction, but if the reactions are not dependent on surface area, the low factor may indicate a “tight” complex. The high factors will usually indicate a “loose” complex [17–19]. Even higher factors (after correction for surface area) can be obtained for complexes having free translation on the surface. Since the concentrations in solids are not controllable in many cases, it would have been convenient if the magnitude of the pre-exponential factor indicated for reaction molecularity. On the basis of these reasons, the thermal decomposition reaction of MgHPO4 · 3H2O may be interpreted as “loose complexes”, which corresponds to the loss of crystallization water and the deprotonated hydrogen phosphate group. These results are consistent with thermal analysis, which confirm that the decomposition product as magnesium pyrophosphate (Mg2P2O7).
Determination of the thermodynamic functions
The calculated values of ∆H*, ∆S* and ∆G* for the decomposition step of MgHPO4 · 3H2O are 98.32 kJ mol−1, −70.03 J K−1 mol−1 and 129.29 kJ mol−1, respectively. The entropy of activation (∆S*) value for the decomposition step is negative. It means that the corresponding activated complexes were with higher degree of arrangement than the initial state. Since the decomposition of MgHPO4 · 3H2O proceeds as a single reaction, the formation of the activated complex passed in situ. In the terms of the activated complex theory (transition theory) [12, 13, 21, 26], a positive value of ∆S* indicates a malleable activated complex that leads to a large number of degrees of freedom of rotation and vibration. A result may be interpreted as a ‘‘fast’’ stage. On the other hand, a negative value of ∆S* indicates a highly ordered activated complex and the degrees of freedom of rotation as well as of vibration is less than they in the non activated complex. This results may indicate a ‘‘slow’’ stage [12, 13, 21, 26]. Therefore, the decomposition reaction of MnHPO4 · 3H2O may be interpreted as ‘‘slow’’ stages. The positive value of the enthalpy ΔH* is in good agreement with an endothermic effects in DTA data. The positive values of ΔH* and ΔG* for the decomposition stage shows that it is connected with the introduction of heat and it is non-spontaneous process. These thermodynamic functions are in consistent with kinetic parameters.
Conclusions
MgHPO4 · 3H2O decomposes in a well-defined step by starting after 323 K and the final product is Mg2P2O7, which is important for its further treatment. The kinetics of the thermal decomposition of MgHPO4 · 3H2O was studied using non-isothermal TG applying model-fitting method. The activation energies calculated for the decomposition step of MgHPO4 · 3H2O by different methods and techniques were found to be consistent, which indicate the independent process and the nature of non-isothermal methods as well as TGA. A correlation between the T p (DTA) and the wavenumber of activated complex assigned to the breaking bond is possible, which can be used for identification in each thermal transition step of the studied compound and will be an alternative method for the calculated wavenumbers of interesting materials. On the basis of correctly established values of the apparent activation energy, pre-exponential factor and the changes of entropy, enthalpy and Gibbs free energy, certain conclusions can be made concerning the mechanisms and characteristics of the process, which play an important role in theoretical study, application development and industrial production of a compound as a basis of theoretical. Consequently, these data will be important for further studies of the studied compound and are applied to solve various scientific and practical problems involving the participation of solid phases.
References
Mesíková Ž, Šulcová P, Trojan M. Synthesis and characterization of newberyite. J Therm Anal Cal. 2007;88:103–6.
Aramendía MA, Borau V, Jiménez C, Marinas JM, Romero FJ. Synthesis and characterization of magnesium phosphates and their catalytic properties in the conversion of 2-hexanol. J Colloid Interface Sci. 1999;217:288–98.
Boonchom B, Maensiri S, Danvirutai C. Soft solution synthesis, non-isothermal decomposition kinetics and characterization of manganese dihydrogen phosphate dihydrate Mn(H2PO4)2·2H2O and its thermal transformation products. Mater Chem Phys. 2008;109:404–10.
Wang C-M, Liau H-C, Tsai W-T. Effect of heat treatment on the microstructure and electrochemical behavior of manganese phosphate coating. Mater Chem Phys. 2007;102:207–13.
Wang C-M, Liau H-C, Tsai W-T. Effects of temperature and applied potential on the microstructure and electrochemical behavior of manganese phosphate coating. Surf Coat Technol. 2006;210:2994–3001.
Vlase T, Vlase G, Birta N, Doca N. Comparative results of kinetic data obtained with different methods for complex decomposition steps. J Therm Anal Cal. 2007;88:631–5.
Ozawa T. A new method of analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38:1881–6.
Kissinger HE. Reaction Kinetics in differential thermal analysis. J Anal Chem. 1957;29:1702–6.
Akahira T, Sunose T. Res Report Chiba Inst Technol (Sci Technol). 1971;16:22–31.
Vlaev L, Nedelchev N, Gyurova K, Zagorcheva M. A comparative study of non-isothermal kinetics of decomposition of calcium oxalate monohydrate. J Anal Appl Pyrolysis. 2008;81:253–62. Reference therein.
Coats AW, Redfern JP. Kinetic parameters from thermogravimetric data. Nature. 1964;20:68–9.
Ŝesták J. Thermodynamical properties of solids. Prague: Academia; 1984.
Young D. Decomposition of solids. Oxford: Pergamon Press; 1966.
Vlase T, Vlase G, Doca M, Doca N. Specificity of decomposition of solids in non-isothermal conditions. J Therm Anal Cal. 2003;72:597–604.
Vlaev LT, Nikolova MM, Gospodinov GG. Non-isothermal kinetics of dehydration of some selenite hexahydrates. J Solid State Chem. 2004;177:2663–9.
Gao X, Dollimore DA. The thermal decomposition of oxalates. Part 26: a kinetic study of the thermal decomposition of manganese (II) oxalate dihydrate. Thermochim Acta. 1993;215:47–63.
Gabal MA. Kinetics of the thermal decomposition of CuC2O4−ZnC2O4 mixture in air. Thermochim Acta. 2003;402:199–208.
Zhang K, Hong J, Cao G, Zhan D, Tao Y, Cong C. The kinetics of thermal dehydration of copper(II) acetate monohydrate in air. Thermochim Acta. 2005;437:145–9.
Cordes HM. Preexponential factors for solid-state thermal decomposition. J Phys Chem. 1968;72:2185–9.
Criado JM, Pérez-Maqueda LA, Sánchez-Jiménez PE. Dependence of the preexponential factor on temperature. J Therm Anal Cal. 2005;82:671–5.
Boonchom B. Kinetics and thermodynamic properties of the thermal decomposition of manganese dihydrogenphosphate dihydrate. J Chem Eng Data. 2008;53:1553–8.
Herzberg G. Molekülspektren und Molekülstruktur. I. Zweiatomige Moleküle. Dresden: Steinkopff; 1939.
Colthup NB, Daly LH, Wiberley SE. Introduction to infrared and Raman spectroscopy. New York: Academic Press; 1964.
Boonchom B, Danvirutai C. A simple synthesis and thermal decomposition kinetics of MnHPO4 center dot H2O rod-like microparticles obtained by spontaneous precipitation route. J Optoelectron Adv Mater. 2008;10:492–9.
Boonchom B, Danvirutai C. Thermal decomposition kinetics of FePO4·3H2O precursor to synthetize spherical nanoparticles FePO4. Ind Eng Chem Res. 2007;46:9071–6.
Boonchom B, Danvirutai C. Synthesis of MnNiP2O7 and nonisothermal decomposition kinetics of a new binary Mn0.5Ni0.5HPO4·H2O precursor obtained from a rapid coprecipitation at ambient temperature. Ind Eng Chem Res. 2008;47:5976–81.
Acknowledgements
The authors thank the Chemistry and Physics Departments, Khon Kaen University for providing research facilities. This work is financially supported by the Thailand Research Fund (TRF) and the Commission on Higher Education (CHE): Research Grant for New Scholar (MRG5280073), Ministry of Science and Technology, Thailand.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Boonchom, B. Kinetic and thermodynamic studies of MgHPO4 · 3H2O by non-isothermal decomposition data. J Therm Anal Calorim 98, 863–871 (2009). https://doi.org/10.1007/s10973-009-0108-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-009-0108-2