
Abstract
Reducing organic groups on hydrophobic silica aerogels (SA) is worth exploring for lowering their thermal hazard risk. In this work, we used dimethyldichlorosilane (DMDCS) to modify silica alcogels, investigated the effects of DMDCS concentration and focused on the thermal hazard assessment of the DMDCS modified SA (DSA). It was turned out that the DSA had less −CH3 content in spite of the thermal stability close to the trimethylchlorosilane modified SA (TSA), about 240 °C. The kinetics study suggested the apparent activation energy (Ea) could be divided into two segments, corresponding to the two processes in the pyrolysis. The positive enthalpy and entropy changes indicated that the thermal oxidation of the DSA was an exothermic reaction, which could not occur without external energy supply. The average Ea of the DSA was far larger than that of the TSA and the gross calorific value of the DSA decreased by about 12% compared with that of the TSA. All these results drew a conclusion that the DMDCS modified SA reduced the thermal hazard to some degree, which provided one possible solution to further lower the thermal hazard of hydrophobic SA.

Highlights
-
Dimethyldichlorosilane modified silica aerogels (DSA) were prepared at the optimum concentration of 4%.
-
Kinetic and thermodynamic behavior of DSA were studied in detail.
-
DSA has larger apparent activation energy than trimethylchlorosilane modified silica aerogels (TSA).
-
DSA has lower thermal hazard than TSA for a less gross calorific value.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Silica aerogels (SA) have been widely used in various fields, which are a type of mesoporous materials with some excellent physicochemical properties, for instance, a low density of 0.003–0.500 g/cm3, a high porosity of over 90%, a large specific surface area of 500–1200 m2/g, and a low thermal conductivity of 0.01–0.03 W/(m·K) [1, 2]. SA have been proved to be promising materials in various applications like aerospace [3], adsorption [4], catalyst supports [5], and drug delivery and targeting systems [6]. But most importantly, SA can obtain wide usages in the field of thermal insulation because of their excellent thermal insulation performance [7, 8].
For a long time, the primary preparation technology of aerogels is to dry wet gels under supercritical condition [1], e.g., supercritical extraction of ethanol in liquid CO2. However, the high equipment costs, safety problems, and the deficiency of continuously industrial production of supercritical drying limit the wide application of SA. Developing the application of advanced equipment and new green materials is a strong measure to control the hazard [9]. Under the efforts of the scientific community and industry, the preparation technology of aerogels under ambient pressure drying (APD) is gradually becoming popular. The core step for synthesizing ambient pressure dried aerogels is the surface modification, which introduces a certain number of organic functional groups (e.g., alkyl) onto the skeletons of gels [10], leading to a reduction of the capillary pressure. This vital procedure is necessary to avoid the destruction of the three-dimensional network structure of SA during APD. On the other hand, the introduced organic functional groups are flammable [11, 12], which is an unavoidable problem we must consider. For thermal insulation materials, the usage environment is so complicated that insulation materials have to face a high temperature and even external heat flux produced by some readily flammable materials.
In reality, people habitually tend to consider aerogel materials as incombustible materials due to their excellent properties, especially their thermal insulation performance. There is little attention drawn on the fire safety of aerogel materials, which definitely increases the thermal hazard risk of aerogels. As the usage of aerogel materials expands increasingly, this risk is getting worse. In our previous work, we investigated and compared the fire performance of supercritically dried SA and ambient pressure dried SA and demonstrated the flammability of hydrophobic SA [11]. In recent years, Ghazi Wakili et al. reported their research on the reaction of ceramic fiber/aerogels to fire exposure [13]; He and Huang et al. also studied the effects of heat treatment on hydrophobic SA and clarified the variations of O/Si, C/Si values, and the products during pyrolysis process [14]. These researches further verified the thermal hazard risks of hydrophobic SA. At present, it is the exact time for us to take the flammability and thermal degradation of hydrophobic SA into consideration. Hence, the remained problem is for us to find out how to reduce these risks.
Just as mentioned above, it is the organic groups introduced by surface modification that bring the thermal hazards to hydrophobic SA. This conclusion can be strongly supported by many thermal analyses [14,15,16,17], which reveals that the exothermic reactions during the pyrolysis process should be ascribed to the thermal oxidation of organic groups (e.g., −CH3). Obviously, the higher the content of organic groups, the higher the thermal hazard risk of hydrophobic SA. Hence, reducing the content of organic groups without compromising other properties or choosing a more appropriate type of organic functional groups are our directions to solve these problems.
Currently, trimethylsilyl (TMS) has been common functional groups for surface modification of aerogels [18, 19], such as trimethylchlorosilane (TMCS), hexamethyldisiloxane (HMDSO) [20], hexamethyldisilazane (HMDZ), etc. Each TMS is composed of three −CH3 groups. Assuming that for a stable hydrophobicity, the quantity of functional groups on the skeletons of gels is approximately constant, decreasing the number of methyl in the functional groups is of great importance to reduce the thermal hazard risk. As it is known, dimethylsilyl (DMS) has less −CH3 groups to graft onto gels during surface modification compared with TMS. Therefore, the challenge we are facing is whether it is feasible to lower the flammability of hydrophobic SA by replacing TMS groups with DMS groups.
In this study, we primarily focused on investigating the thermal hazard of hydrophobic SA modified with DMS groups. Hence, we first prepared the DMS modified hydrophobic SA and compared them with the classical hydrophobic SA modified by TMS groups on the aspect of basic physicochemical properties. Followed by the thermal analyses, kinetic and thermodynamic analyses were carried out to evaluate their thermal hazard. The research outcome demonstrated that this strategy is beneficial to lower the thermal hazard of hydrophobic SA, which can provide a technical guide on further reducing the flammability of SA.
2 Experimental methodology
2.1 Raw materials and preparation
Tetraethylorthosilicate (TEOS, 98%), dimethyldichlorosilane (DMDCS, 98%), from Aladdin, China, were used as the precursor and surface modifier. Other chemicals including ethanol (EtOH), TMCS, n-hexane, hydrochloric acid (HCl), and ammonium hydroxide (NH3·H2O) were chemical pure grade, which were purchased from Sinopharm Chemical Reagent Co., Ltd. (SCRC, China).
The acid-base catalyzed sol–gel process and derivatization modification method were used to prepare hydrophobic SA in this work. The optimum molar ratio of TEOS: EtOH: H2O: HCl: NH3·H2O was fixed at 1: 9.6: 2.2: 1.6 × 10−3: 9.7 × 10−3 referring to our previous work [21]. The first step was to mix TEOS, EtOH, DI H2O, and 0.1 M HCl together and stir for 10 min in a 100 mL beaker. After completing the hydrolysis in a 45 °C water bath for 9 h, 0.5 M NH3·H2O was added in the mixed sol and stirred for 5 min. The gelation usually occurred within 30 min after the alcogels were made airtight for 4 h for strengthening the gel network. After aging, the alcogels were placed in n-hexane at 45 °C for solvent exchange. The alcogels were cut into small pieces and were immersed in a mixture of DMDCS/n-hexane solvent to perform the surface modification, during which the percentage of DMDCS in the solvent was elevated from 2 to 10%. Finally, the wet alcogels were washed in n-hexane for several times and dried at 120 °C for 4 h under ambient pressure to get hydrophobic SA in granular form. In this work, the as-prepared DMDCS modified SA (DSA) and TMCS modified SA were denoted as DSA and trimethylchlorosilane modified SA (TSA), respectively.
2.2 Methods of characterization
The bulk density (ρb) of SA was measured by the Archimedes principle. Specifically, the SA was pressed into deionized water to measure volume displacement by using an ultrafine steel wire. The porosity was determined by Eq. (1) [22]:
where ρs is the skeletal density of SA, usually about 2200 kg/m3 [15, 17, 23]. The pH was monitored by accurate pH test papers ranging between 0.5 and 5.0 (Newstar, China). The thermal conductivity instrument (TC3000E, XIATECH, China) based on transient hot-wire method was used to measure the thermal conductivity at 25 °C. First, the SA samples were ground into fine powders with 200 mesh sieve and then the kapton sensor was insert into SA powders through the slit on the stainless steel sample holder with 500 g weight on silica aerogel powders. For thermal conductivity measurement, each sample was tested over three times and the averages were taken as the final results. The microstructures were observed using a double beam microscope (Helios Nanolab G3UC, FEI). Furthermore, the hydrophobicity was characterized by a contact angle meter (JC2000D1, Shanghai Zhongchen Instrument) through putting a water droplet of 5 μL on the sample surface and the contact angles were acquired by the image processing program, ImageJ [24].
The Quadrasorb SI-3MP analyzer (Quantachrome, USA) was used to test the N2 adsorption–desorption isotherms at 77 K with three specimens for each recipe. The specific surface area and pore size distribution (PSD) were calculated using the Brunauer–Emmett–Teller method [25] and Barrette–Joynere–Halenda method from the desorption branch [26], respectively. The chemical bonds were studied by Fourier transform infrared spectroscopy, which was tested by a Nicolet iS50 (Thermo Fisher Scientific, USA) by pressing silica aerogel pieces in a KBr pellet and collecting the spectra from 400 to 4000 cm−1.
Thermogravimetric analysis (TG, STA 449F3, NETZSCH) was carried out at three different heating rates, i.e., 10, 15, 25 °C/min, from room temperature to 1000 °C in air to investigate the thermal stability, kinetics, and thermodynamic analyses of SA [27–30]. The temperature resolution is 0.001 K and the balance resolution is 0.1 μg. An oxygen bomb calorimeter (C3000, IKA, Germany) was used to measure the gross calorific value (GCV) following the ISO 1716: 2010 standard. It should be pointed out that the error bars used in this study are standard errors and over three specimens were tested for the calculation of the average values and standard errors.
2.3 Kinetics and thermodynamic analysis
The kinetic equation of chemical reaction can be written as Eq. (2).
in which, α is the conversion rate and can be calculated as
where mi, mα, and m∞ are the initial mass, the mass at α, and the finally residual mass, respectively. f(α) is a reaction model [31], which reflects the reaction mechanism. k(T) is reaction rate constant and is usually defined as
where A and Ea is a preexponential factor and apparent activation energy, respectively. R is the universal gas constant, 8.314 J/(K·mol). Therefore, the fundamental expression of the analytical method to calculate kinetic parameters can be described as follows.
Under nonisothermal conditions with a constant heating rate, the temperature can be expressed as,
where β is the heating rate and T0 is the initial temperature.
After combining Eq. (6), the conversion rate can be transformed into a temperature-derivative, i.e.,
Rearrange Eq. (7) to separate variables α and T, and then integrate it.
2.3.1 Flynn–Wall–Ozawa (FWO) method
In the FWO method, after introducing Doyle’s approximation, it can obtain the linear form [32, 33].
For various heating rates at a fixed conversion rate α, the lnβ and the corresponding 1/T can be fitted to a straight line. Finally, the apparent Ea can be calculated from the slope, −1.052E/R.
2.3.2 Kissinger–Akahira–Sunose (KAS) method
The following equation is employed in KAS method [34].
At each degree of conversion α, the ln(β2/T) and l/T data points obtained from different heating rates were fitted to a straight line. Then the Ea can be calculated by the slope, −E/R.
2.3.3 Thermodynamic analysis
Based on obtained apparent activation energy, the thermodynamic parameters including the pre-exponential factor (A), the change of enthalpy (ΔH), free Gibbs energy (ΔG), and entropy (ΔS) can be calculated as per the following equations [35]:
where Tm and Tα are the peak temperature on DTG and the temperature at a conversion of α, respectively; kB and h are Boltzmann constant (1.381 × 10−23 J/K) and Plank constant (6.626 × 10−34 J·s), respectively.
3 Results and discussion
3.1 Physicochemical properties
The pictures of the unmodified xerogels and the DSA with different DMDCS concentration are presented in Fig. 1, in which the DSA with the DMDCS concentration at 4–6% shows light blue, indicating a better surface modification. Figure 2 compares the density, porosity, and thermal conductivity of the prepared DSA modified with different concentration from 2 to 10%. It is obvious that the DSA at the DMDCS concentration of 4% has the lowest density and thermal conductivity and the highest porosity. When the DMDCS concentration increases from 4 to 10%, the density and thermal conductivity increase slightly while the porosity decreases. The relatively larger thermal conductivity of the DSA should be caused by the compaction (500 g weight) during the measurement and unoptimized preparation parameters.

Pictures of the unmodified xerogels and the DSA (0.5 g) with various DMDCS concentration between 2 and 10%

The changes of a density, porosity, and b thermal conductivity of DSA
During the modification, the wet gels were immersed into n-hexane/DMDCS mixture solvent in a sealed breaker. With the surface modification proceeding, the derived by-product, i.e., HCl, was continuously generated in the mixture solvent, which resulted in an acid environment. As the DMDCS concentration increases, more HCl is generated and lead to a decreasing pH, as listed in Table 1. In an overly acid mixture solvent, the surface modification is hard to proceed because a large number of H+ in the acidic solvent can hinder the reaction between DMDCS and the Si–OH groups on the wet gels due to chemical equilibrium. Besides, because of the two reactive sites, DMDCS may react with themselves, which results in a part of DMDCS losing the efficacy of surface modification. Thus, the appropriate concentration of DMDCS is vital for high-efficiency surface modification.
The typical microstructures of as-prepared DSA and the unmodified xerogels are presented in Fig. 3, which shows the influence of the modification treatment in different DMDCS concentrations on the DSA. In Fig. 3b it can be seen that the DSA modified in a DMDCS concentration being 4% displays a more porous structure compared with other DMDCS concentrations. When the DMDCS concentration is <4% or >6%, the pore size decreases significantly and a denser microstructure appears. This can be explained as follows: When the DMDCS concentration is <4% as presented in Fig. 3a, the number of Si–CH3 groups is so less that the gel surface cannot acquire an adequate modification; while the DMDCS concentration is over 6% as shown in Fig. 3c, d, the overly acidic environment restrains the surface modification as mentioned above. Besides, DMDCS render themselves at high concentration and readily react with each other, which also impedes the surface modification. All of these cases result in the shrinkage or even destruction during APD. The variation of the microstructure of DSA exactly corresponds to the change of the density.

Microstructures of DSA modified with various DMDCS concentration, a 2%, b 4%, c 6%, d 10%, and e unmodified xerogels
Figure 4a compares the N2 adsorption–desorption isotherms of the DSA modified by different DMDCS concentrations and the PSD is presented in Fig. 4b. Obviously, all the samples exhibit typical type IV isotherms and the hysteresis loops of type H3, which indicate the attribution of mesoporous materials and the existence of slit-like pores [36]. The detailed pore parameters are listed in Table 2 with the typical specific surface area of more than 850 m2/g. Furthermore, corresponding to the microstructure, the average pore size of DSA modified at the DMDCS concentration of 4% is larger than the others.

a N2 adsorption–desorption isotherms and b PSD of the DSA modified with different DMDCS concentration
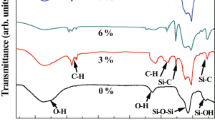
In contrast to the FTIR spectra of the unmodified xerogels, which is shown from Fig. 5, the FTIR spectra of the DSA modified by different DMDCS concentration are almost the same. The outstanding peak with the absorption spectra around 1090 cm−1 is corresponding to the asymmetric flexible vibration mode of Si–O–Si bond [21], while the inconspicuous peaks at 800 and 460 cm−1 belongs to the symmetric vibration and bending vibration of Si–O–Si bond. The absorption spectra around 2954 and 1398 cm−1 just correspond to the C–H bond and the Si–C bond appears around 1265 and 845 cm−1 [37]. The existence of these characteristic functional groups indicates a successful surface modification [38]. Compared with unmodified xerogels, the −OH intensities were significantly reduced, which also implies that the organic groups have been grafted on the skeleton of the DSA with the replacement of Si–OH groups. These introduced organic groups exactly constitute the chemical foundation of the hydrophobicity of the DSA.

The FTIR spectra of unmodified silica xerogels and DSA modified by different concentrations of DMDCS
Furthermore, the hydrophobicity of DSA has been confirmed by the water contact angle as shown in Fig. 6. Obviously, the maximum contact angle appears at 4% DMDCS concentration and the value is 154°. This result is consistent with our former analyses to some extent.

a The change of the water contact angle along with the DMDCS concentration and b the photograph of contact angle at the DMDCS concentration being 4%
3.2 Thermostability analysis
As shown in Fig. 7a, b, the TG curves of DSA and TSA both have a slight weight before 150 °C, indicating the evaporation of water and residual organic solvents [17]. Furthermore, the sharp weight losses from 250 to 400 °C on the TG curves are accompanied by obvious exothermic peaks on the DSC curves. These processes are commonly attributed to the thermal oxidative decomposition of Si–CH3 groups on the skeletons [12].

TG-DSC curves of a the DSA and b TSA under 10 °C/min in air
In term of a thermal reaction process, the onset temperatures and the temperatures corresponding to the fastest reaction rate are two important parameters to determine the thermal stability of a material, which are usually denoted as Tonset and Tpeak, respectively. For hydrophobic SA, when the temperature exceeds Tonset, SA becomes hydrophilic gradually, resulting in adsorption of moisture in air and further causing a continuous deterioration of physical properties.
As seen in Table 3, the Tonset of the DSA and TSA are 238 and 242 °C, respectively, while the Tpeak of the DSA and TSA are 265 and 273 °C, respectively. Comparing the Tonset and Tpeak between the DSA and TSA, it can draw a conclusion that they have close thermal stability. Furthermore, the weight losses from the onset temperature to the end temperature during the exothermic reaction can be approximately regarded as the −CH3 content on SA. Accordingly, the −CH3 contents of DSA and TSA are calculated as about 6 and 9% through the TG curves, respectively. This demonstrates that the DSA possesses about 33% less −CH3 groups than that of the TSA. From the perspective of the combustible component, the DSA further presents a lower thermal hazard compared with the TSA.
3.3 Kinetic analysis
For conversion rate α within 0.1 ~ 0.9, the two methods, i.e., FWO and KAS methods, are employed to calculate the apparent activation energy Ea. The optimum fitting lines are confirmed by the least square method. Within α = 0.1–0.9, the fitted lines for ln(β) ~ 1/T in Fig. 8a and ln(β/T2) ~ 1/T in Fig. 8b all present high linearity. Finally, the apparent activation energies are calculated according to the slopes of the corresponding fitted lines.

Fitted lines based on a the OFW method and b the KAS method
The variations of the apparent activation energy are presented in Fig. 9, which indicates that the apparent activation energies calculated from the FWO method and the KAS method are almost the same. Note that the apparent activation energies calculated within α < 0.2 and α > 0.8 are not reliable. Because the evaporation of some remained solvent in the initial stage and some residue are no longer decomposed in the final stage therefore it may cause an error in calculating kinetic parameters [39]. Without considering the initial and final stage of the thermal oxidation, namely defining α = 0.2–0.8, the conspicuous two-segment process is presented and the cut-off point is set as α = 0.5, which declares the thermal oxidation processes for hydrophobic DSA are multistage.

Ea along the increasing α for the DSA
In Stage I, namely α = 0.2–0.5, the change of Ea shows an obvious increasing trend with a higher α and the Ea rises rapidly to the maximum, 257.1 kJ/mol. This stage mainly corresponds to the decomposition of organic groups on skeletons. As the thermal oxidation of Si–CH3 groups proceeds, more Si–OH groups are generated [11, 14], which can be expressed as Eq. (15).
Simultaneously, the condensation reaction among the produced Si–OH groups would launch and generate more water in the process subsequently. Accompanied by these reactions, the E increases rapidly [40] and rises to the maximum at α = 0.5. In Stage II within α = 0.5–0.8, Ea decreases promptly, which indicates that certain intermediates may facilitate the decomposition and oxidation of organic groups [11]. As it is known to all, the Ea reflects an energy obstacle that must be overcome for proceeding a chemical reaction. Therefore, a higher value of Ea implies it is more difficult for a chemical reaction to occur.
3.4 Thermodynamic analysis
Table 4 shows the thermodynamic parameters including pre-exponential factor (A), the changes of enthalpy (ΔH), free Gibbs energy (ΔG), and entropy (ΔS), which are calculated by using the average Ea obtained from the two methods.
The values of the pre-exponential factor (A) range from 8.2 × 106 to 4.2 × 1022, which are directly related to the material structure to some extent. Besides, it can also be used to describe the complex reactions that occur during decomposition [41]. Within α = 0.2–0.5 for Stage I, the values of A increase rapidly indicating a higher rate of molecular collisions and an increasing need for energy during the pyrolysis of the DSA [42]. On the contrary, the values of A decrease sharply at stage II, which just keep consistent with the change of apparent activation energy.
The change of enthalpy (ΔH) reflects the energy difference between the reactants and products, which nearly has the same change with the Ea. Furthermore, all the ΔH is larger than zero indicates an exothermic reaction of the pyrolysis process. The change of Gibbs free energy (ΔG) means the total increase in energy of the reaction system for the reagents and the formation of the activated complex [35, 42,43,44]. During the pyrolysis process of the DSA, the ΔG has no obvious changes with a slight fluctuation from 156 to 161 kJ/mol. In other words, the total energy required in the pyrolysis process has no significant difference in the whole process and the whole pyrolysis process cannot be carried out spontaneously without external energy supply. The entropy (ΔS) represents the disorder degree of a system, which can be expressed as a state parameter [42]. As listed in Table 4, the ΔS ranges within −126 to 175 J/K/mol. The negative values indicate that the degree of disorder of products is lower compared with the initial reactants due to the external energy supply [42, 43, 45], and vice versa.
3.5 Thermal hazard assessment
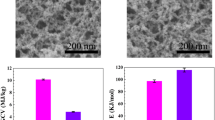
The GCV can reflect the total heat release of a material in the condition of complete combustion precisely [46,47,48,49,50], which usually measured using an oxygen bomb calorimeter. As illustrated in Fig. 10, the GCV of the TSA is 10.1 MJ/kg while that of DSA is 8.9 MJ/kg. Compared with the TSA, the DSA possesses about 12% reduction in GCV. Furthermore, the average apparent activation energy of the TSA and DSA is calculated as 97 ± 18 and 174 ± 42 kJ/mol, respectively. That is to say, the DSA are harder to activate the thermal oxidation process compared with the TSA.

GCV of the TSA and DSA
Here, we further give a more comprehensive thermal hazard assessment on the DSA. From the TG–DSC analyses, the DSA nearly has the same thermal stability as the typical TSA, which has been indicated by the close Tonset and Tpeak. The specific thermodynamic parameters have demonstrated that the whole pyrolysis process of the DSA cannot occur spontaneously without external energy supply. The apparent activation energy of the DSA is far larger than that of the TSA, which indicates that more energy is needed to launch the thermal oxidation reaction of the DSA. Furthermore, a more direct characterization for thermal hazard is carried out by the GCV measurement, which has clarified that the DSA possesses a lower GCV than the TSA. This result can be supported by the reduction in −CH3 content for the TSA. All of these indicate that the DSA has better fire retardancy than the TSA. In one word, it can be concluded that the DSA has a lower potential thermal hazard compared to the TSA.
4 Conclusions
In this study, DMDCS modified hydrophobic silica aerogels (DSA) were prepared successfully. The effects of DMDCS concentration were investigated and the optimum was confirmed as 4%. Based on the as-prepared DSA, we focused on the assessment of their thermal hazards and some conclusions were drawn as follow.
The TG–DSC analyses indicated that the DSA had similar thermal stability with the TSA but possessed less −CH3 content compared with the TSA. The cut-off point was set at α = 0.5, which divided the whole thermal oxidation process into two stages, i.e., α = 0.2–0.5 for Stage I and α = 0.5–0.8 for Stage II. Besides, the average apparent activation energy of the DSA was far larger than that of the TSA, which implied that more energy was required to launch the thermal oxidation reaction. On the aspect of thermodynamics, all the ΔH were larger than zero during the pyrolysis, indicated the attribution of an exothermic reaction. The ΔG fluctuating slightly within 156–161 kJ/mol demonstrated that the thermal oxidation of the DSA cannot occur spontaneously without external energy supply. Furthermore, the GCV of the DSA was reduced by about 12% compared with that of the TSA. All these kinetic and thermodynamic parameters revealed that the DSA lowered the thermal hazard to some extent, which provided a technical basis for further reducing the thermal hazards of hydrophobic SA.
References
Husing N, Schubert U (1998) Aerogels-airy materials: chemistry, structure, and properties. Angew Chem Int Ed Engl 37:22–45. https://doi.org/10.1002/(SICI)1521-3773(19980202)37:1/2<22::AID-ANIE22>3.0.CO;2-I
Cuce E, Cuce PM, Wood CJ, Riffat SB (2014) Toward aerogel based thermal superinsulation in buildings: a comprehensive review. Renew Sustain Energy Rev 34:273–299. https://doi.org/10.1016/j.rser.2014.03.017
Randall JP, Meador MAB, Jana SC (2011) Tailoring mechanical properties of aerogels for aerospace applications. Acs Appl Mater Interfaces 3:613–626. https://doi.org/10.1021/am200007n
Rao AV, Hegde ND, Hirashima H (2007) Absorption and desorption of organic liquids in elastic superhydrophobic silica aerogels. J Colloid Interface Sci 305:124–132. https://doi.org/10.1016/j.jcis.2006.09.025
Zhao Y, Liang Y, Zhao X et al. (2011) Preparation and microstructure of CuO-CoO-MnO/SiO2 nanocomposite aerogels and xerogels as catalyst carriers. Prog Nat Sci Mater Int 21:330–335. https://doi.org/10.1016/S1002-0071(12)60065-3
Alnaief M, Smirnova I (2010) Effect of surface functionalization of silica aerogel on their adsorptive and release properties. J Non-Cryst Solids 356:1644–1649. https://doi.org/10.1016/j.jnoncrysol.2010.06.027
Koebel M, Rigacci A, Achard P (2012) Aerogel-based thermal superinsulation: an overview. J Sol–Gel Sci Technol 63:315–339. https://doi.org/10.1007/s10971-012-2792-9
Baetens R, Jelle BP, Gustavsen A (2011) Aerogel insulation for building applications: a state-of-the-art review. Energy Build 43:761–769. https://doi.org/10.1016/j.enbuild.2010.12.012
Wang B, Wu C, Kang L et al. (2018) Work safety in China’s thirteenth five-year plan period (2016–2020): current status, new challenges and future tasks. Saf Sci 104:164–178. https://doi.org/10.1016/j.ssci.2018.01.012
Mahadik SA, Pedraza F, Parale VG, Park H-H (2016) Organically modified silica aerogel with different functional silylating agents and effect on their physico-chemical properties. J Non-Cryst Solids 453:164–171. https://doi.org/10.1016/j.jnoncrysol.2016.08.035
Li Z, Cheng X, Shi L et al. (2016) Flammability and oxidation kinetics of hydrophobic silica aerogels. J Hazard Mater 320:350–358. https://doi.org/10.1016/j.jhazmat.2016.07.054
Li Z, Huang S, Shi L et al. (2019) Reducing the flammability of hydrophobic silica aerogels by doping with hydroxides. J Hazard Mater 373:536–546. https://doi.org/10.1016/j.jhazmat.2019.03.112
Ghazi Wakili K, Remhof A (2017) Reaction of aerogel containing ceramic fibre insulation to fire exposure. Fire Mater 41:29–39. https://doi.org/10.1002/fam.2367
He S, Huang Y, Chen G et al. (2019) Effect of heat treatment on hydrophobic silica aerogel. J Hazard Mater 362:294–302. https://doi.org/10.1016/j.jhazmat.2018.08.087
Zhang W, Li Z, Shi L et al. (2019) Methyltrichlorosilane modified hydrophobic silica aerogels and their kinetic and thermodynamic behaviors. J Sol–Gel Sci Technol 89:448–457. https://doi.org/10.1007/s10971-018-4882-9
Luo Y, Li Z, Zhang W et al. (2019) Rapid synthesis and characterization of ambient pressure dried monolithic silica aerogels in ethanol/water co-solvent system. J Non-Cryst Solids 503:214–223. https://doi.org/10.1016/j.jnoncrysol.2018.09.049
Li Z, Cheng X, Gong L et al. (2018) Enhanced flame retardancy of hydrophobic silica aerogels by using sodium silicate as precursor and phosphoric acid as catalyst. J Non-Cryst Solids 481:267–275. https://doi.org/10.1016/j.jnoncrysol.2017.10.053
Malfait WJ, Zhao S, Verel R et al. (2015) Surface chemistry of hydrophobic silica aerogels. Chem Mater 27:6737–6745. https://doi.org/10.1021/acs.chemmater.5b02801
Malfait WJ, Jurányi F, Zhao S et al. (2017) Dynamics of silica aerogel’s hydrophobic groups: a quasielastic neutron scattering study. J Phys Chem C 121:20335–20344. https://doi.org/10.1021/acs.jpcc.7b06011
Iswar S, Griffa M, Kaufmann R et al. (2019) Effect of aging on thermal conductivity of fiber-reinforced aerogel composites: an X-ray tomography study. Microporous Mesoporous Mater 278:289–296. https://doi.org/10.1016/j.micromeso.2018.12.006
Li Z, Cheng X, He S et al. (2015) Characteristics of ambient-pressure-dried aerogels synthesized via different surface modification methods. J Sol–Gel Sci Technol 76:138–149. https://doi.org/10.1007/s10971-015-3760-y
Soleimani Dorcheh A, Abbasi MH (2008) Silica aerogel; synthesis, properties and characterization. J Mater Process Technol 199:10–26. https://doi.org/10.1016/j.jmatprotec.2007.10.060
Malfait WJ, Verel R, Koebel MM (2014) Hydrophobization of silica aerogels: insights from quantitative solid-state NMR spectroscopy. J Phys Chem C 118:25545–25554. https://doi.org/10.1021/jp5082643
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to imageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Brunauer S, Emmett PH, Teller E (1938) Adsorption of gases in multimolecular layers. J Am Chem Soc 60:309–319. https://doi.org/10.1021/ja01269a023
Barrett EP, Joyner LG, Halenda PP (1951) The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J Am Chem Soc 73:373–380. https://doi.org/10.1021/ja01145a126
Wu X, Fan M, Shen X et al. (2018) Silica aerogels formed from soluble silicates and methyl trimethoxysilane (MTMS) using CO2 gas as a gelation agent. Ceram Int 44:821–829. https://doi.org/10.1016/j.ceramint.2017.10.005
Cheng X, Li C, Shi X et al. (2017) Rapid synthesis of ambient pressure dried monolithic silica aerogels using water as the only solvent. Mater Lett 204:157–160. https://doi.org/10.1016/j.matlet.2017.05.107
He S, Sun G, Cheng X et al. (2017) Nanoporous SiO 2 grafted aramid fibers with low thermal conductivity. Compos Sci Technol 146:91–98. https://doi.org/10.1016/j.compscitech.2017.04.021
Gurav JL, Rao AV, Rao AP et al. (2009) Physical properties of sodium silicate based silica aerogels prepared by single step sol–gel process dried at ambient pressure. J Alloy Compd 476:397–402. https://doi.org/10.1016/j.jallcom.2008.09.029
Jiang L, Zhang D, Li M et al. (2018) Pyrolytic behavior of waste extruded polystyrene and rigid polyurethane by multi kinetics methods and Py-GC/MS. Fuel 222:11–20. https://doi.org/10.1016/j.fuel.2018.02.143
Flynn JH, Wall LA (1966) A quick, direct method for the determination of activation energy from thermogravimetric data. J Polym Sci Part C Polym Lett 4:323–328
Doyle CD (2010) Estimating isothermal life from thermogravimetric data. J Appl Polym Sci 6:639–642. https://doi.org/10.1002/app.1962.070062406
Ozawa T (1992) Estimation of activation energy by isoconversion methods. Thermochim Acta 203:159–165. https://doi.org/10.1016/0040-6031(92)85192-X
Domingos Maia AA, de Morais LC (2016) Kinetic parameters of red pepper waste as biomass to solid biofuel. Bioresour Technol 204:157–163. https://doi.org/10.1016/j.biortech.2015.12.055
Rojas F, Kornhauser I, Felipe C et al. (2002) Capillary condensation in heterogeneous mesoporous networks consisting of variable connectivity and pore-size correlation. Phys Chem Chem Phys 4:2346–2355. https://doi.org/10.1039/b108785a
Gurav JL, Rao AV, Rao AP et al. (2009) Physical properties of sodium silicate based silica aerogels prepared by single step sol-gel process dried at ambient pressure. J Alloy Compd 476:397–402. https://doi.org/10.1016/j.jallcom.2008.09.029
Rao AP, Rao AV, Pajonk GM (2007) Hydrophobic and physical properties of the ambient pressure dried silica aerogels with sodium silicate precursor using various surface modification agents. Appl Surf Sci 253:6032–6040. https://doi.org/10.1016/j.apsusc.2006.12.117
Kim YS, Kim YS, Kim SH (2010) Investigation of thermodynamic parameters in the thermal decomposition of plastic waste-waste lube oil compounds. Environ Sci Technol 44:5313–5317. https://doi.org/10.1021/es101163e
Zhuravlev LT (2000) The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf Physicochem Eng Asp 173:1–38. https://doi.org/10.1016/S0927-7757(00)00556-2
Turmanova SC, Genieva SD, Dimitrova AS, Vlaev LT (2008) Non-isothermal degradation kinetics of filled with rise husk ash polypropene composites. Express Polym Lett 2:133–146. https://doi.org/10.3144/expresspolymlett.2008.18
Yuan X, He T, Cao H, Yuan Q (2017) Cattle manure pyrolysis process: kinetic and thermodynamic analysis with isoconversional methods. Renew Energy 107:489–496. https://doi.org/10.1016/j.renene.2017.02.026
Xu Y, Chen B (2013) Investigation of thermodynamic parameters in the pyrolysis conversion of biomass and manure to biochars using thermogravimetric analysis. Bioresour Technol 146:485–493. https://doi.org/10.1016/j.biortech.2013.07.086
Sheng J, Ji D, Yu F et al. (2014) Influence of chemical treatment on rice straw pyrolysis by TG-FTIR. Ieri Procedia 8:30–34. https://doi.org/10.1016/j.ieri.2014.09.006
Kim YS, Kim YS, Kim SH (2010) Investigation of thermodynamic parameters in the thermal decomposition of plastic waste-waste lube oil compounds. Environ Sci Technol 44:5313–5317. https://doi.org/10.1021/es101163e
Mancini M, Rinnan A, Pizzi A, Toscano G (2018) Prediction of gross calorific value and ash content of woodchip samples by means of FT-NIR spectroscopy. Fuel Process Technol 169:77–83. https://doi.org/10.1016/j.fuproc.2017.09.021
Mesroghli Sh, Jorjani E, Chehreh Chelgani S (2009) Estimation of gross calorific value based on coal analysis using regression and artificial neural networks. Int J Coal Geol 79:49–54. https://doi.org/10.1016/j.coal.2009.04.002
Patel SU, Kumar BJ, Badhe YP et al. (2007) Estimation of gross calorific value of coals using artificial neural networks. Fuel 86:334–344. https://doi.org/10.1016/j.fuel.2006.07.036
Yamazaki I, Mimuro M, Murao T et al. (1984) Excitation energy transfer in the light harvesting antenna system of the red ALGA Porphyridium cruentum and the blue-green ALGA Anacystis nidulans: analysis of time-resolved fluorescence spectra. Photochem Photobiol 39:233–240. https://doi.org/10.1111/j.1751-1097.1984.tb03432.x
Feng Q, Zhang J, Zhang X, Wen S (2015) Proximate analysis based prediction of gross calorific value of coals: a comparison of support vector machine, alternating conditional expectation and artificial neural network. Fuel Process Technol 129:120–129. https://doi.org/10.1016/j.fuproc.2014.09.001
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51904336), the Fundamental Research Funds for the Central Universities (Nos. 202501003 and 202045001) and the China Scholarship Council (No. 201806375007). Furthermore, we really appreciate the anonymous reviewer for the constructive comments and the co-editor Florence Babonneau for the resubmission opportunity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Y., Li, Z., Huber, L. et al. Reducing the thermal hazard of hydrophobic silica aerogels by using dimethyldichlorosilane as modifier. J Sol-Gel Sci Technol 93, 111–122 (2020). https://doi.org/10.1007/s10971-019-05170-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-019-05170-5