
Abstract
Sulfated zirconia catalysts were prepared using the sol–gel process. Hydrolysis water was produced in situ. The effects of the zirconium precursor on the physicochemical properties and the acidity of the solids have been investigated. The prepared catalysts were characterized by XRD, Raman spectroscopy, XPS, FTIR of adsorbed pyridine, physisorption of N2 at 77 K, and TPR–H2–MS. Pt-promoted catalysts were tested for n-hexane isomerization reaction and the influence of Pt amount on the stability was studied. Obtained solids exhibit low specific surface areas and narrow pore size distributions. In addition, the metastable tetragonal phase of ZrO2 was stabilized at a relatively high temperature. The acidity of catalysts prepared from different zirconium alkoxides and the magnitude of interaction between zirconia and sulfate species are compared and discussed.

Sulfated zirconia catalysts were prepared using the sol–gel process. Hydrolysis water was produced in situ. The effects of the zirconium precursor have been investigated. The prepared catalysts were characterized by XRD, Raman spectroscopy, XPS, FTIR of adsorbed pyridine, physisorption of N2 at 77 K, and TPR–H2–MS. Pt-promoted catalysts were tested for n-hexane isomerization reaction.
Highlights
-
The metastable tetragonal phase of ZrO2 is stabilized at 833 K.
-
The initial catalytic activity of sulfated zirconia is closely related to the density of Lewis acid sites.
-
The use of zirconium propoxide leads to better retention of sulfate groups.
-
The increase in the amount of Pt leads to a greater loss of sulfur.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The catalytic conversion of hydrocarbons through reactions such as alkylation, cracking, or isomerization is a very interesting research area, especially in the petrochemical industry. For instance, the isomerization process, involving the transformation of linear alkanes into their branched counterparts in order to increase the octane number, has become, over the last few years, a promising alternative as it improves the fuel quality in accordance with the new environmental standards [1, 2]. However, the isomerization reactions require the presence of a strong acid catalyst, which should preferentially be solid, so that problems caused by liquid acid catalysts are avoided. In fact, liquid acids are responsible for the corrosion of the reactors. Their separation from the reaction medium is not easy. Several research teams have investigated the development of solid catalysts which would exhibit very strong acidities. Among the different studied solids, sulfated zirconia, discovered by Hino and Arata [3] was shown to be one of the most efficient catalysts. In fact, sulfated zirconia has remarkable acid properties and is able to convert light alkanes into branched counterparts, at a relatively low temperature, which is thermodynamically favorable to the production of isomers. Several studies have been undertaken on this type of catalysts and a special attention was given to its industrial application for conversion of linear alkanes into branched molecules which exhibit high octane numbers [4,5,6,7,8]. Meanwhile, sulfated zirconia catalysts have been tested in different reactions such as alkylation, acylation, esterification, polymerization, and nitration [9,10,11,12,13,14]. The overall results showed that sulfated zirconia solids reveal a high acidity and also very interesting catalytic properties [15,16,17]. However, many studies showed that the characteristics of sulfated zirconia catalysts depend heavily on the preparation conditions, the nature of the zirconium and sulfate precursors, the sulfate loading, and the calcination temperature [18,19,20,21,22,23].
Regarding the great impact of the preparation method on the physicochemical properties of sulfated zirconia samples, it has been reported that the sol–gel process represents a particularly attractive synthetic route of these catalysts [24, 25]. The solids produced by sol–gel process exhibited generally high specific surface areas, a relatively large porosity, and a good homogeneity. In addition, this method offers an interesting flexibility of preparation parameters [26, 27].
In order to develop a very active catalyst, we have shown in previous studies [28, 29] that the decrease in the rate of hydrolysis and condensation steps of zirconium alkoxides, during preparation of sulfated zirconia in a single step by the sol–gel method, is of particular importance. Therefore, instead of adding water directly to the alcoholic solution of the zirconium precursors, the needed water molecules, for the hydrolysis of zirconium alkoxides, were produced in situ. This relatively slow process involves the alcohol dehydration reaction which takes place in the presence of concentrated sulfuric acid. The physicochemical properties of the sulfated zirconia catalysts obtained directly by this technique and particularly the effect of the zirconium alkoxide should be investigated.
Based on this sol–gel preparation process, the main objective of the present study is to compare the structural, textural, and catalytic properties of a series of sulfated zirconia samples prepared using different zirconium alkoxides. Furthermore, the study establishes a relation between acidic properties and the preparation method.
The catalytic performances of the prepared sulfated zirconia catalysts have been evaluated using n-hexane isomerization reaction at relatively low temperatures (443–473 K). In addition to its industrial interest, this reaction was considered as one of the reference tests used to detect the existence of a strong acidity. Furthermore, in order to prevent fast deactivation of the catalyst, due to the blockage of active sites by coke, this reaction requires at the same time the presence of acidic and metallic sites on the surface of the sulfated zirconia catalysts. Therefore, platinum was used, and the effects of its amount on the physicochemical properties and catalytic performances of the prepared sulfated zirconia catalysts were studied.
2 Experimental
2.1 Catalyst preparation
Sulfated zirconia samples were prepared by sol–gel process using three different zirconium alkoxide precursors supplied by Acros. Concentrated H2SO4 (94–96%) was used as a sulfating agent and at the same time as a catalyst for the alcohol dehydration reaction in order to produce, in situ, the water molecules \(2\,{\mathrm {ROH}}\,\underrightarrow {\mathrm{H}_2{\mathrm{SO}_4}}{\mathrm {zROR}} +{\mathrm {H}_2{\mathrm{O}}}\) needed for the hydrolysis of zirconium alkoxide.
In the three cases under study, the preparation process was as follows. First, the desired amount of zirconium precursor was dissolved in the corresponding alcohol in order to obtain an alcoholic solution where the zirconium concentration is fixed at [Zr] = 1 molL−1. After stirring the solution for 30 min, sulfuric acid was added dropwise until achieving a S/Zr molar ratio of 0.5. It should be noted that this ratio was selected as most research groups interested in sulfated zirconia catalysts have shown that it was an optimal ratio [30, 31]. Transparent gels obtained after a relatively long time (more than 10 days), were then dried at 393 K for 24 h. Finally, all samples were calcinated in a quartz cell at 833 K, under an oxygen flow of 30 cm3min−1. Synthesized catalysts were noted SZP, SZB, and SZtB when zirconium propoxide (70% in propanol), zirconium butoxide (80% in butanol), and zirconium tert-butoxide were used as zirconium precursors, respectively.
2.2 Catalyst characterization
2.2.1 XRD
Powder XRD analysis was performed using a PHILIPS analytical diffractometer using Cu Kα radiation (λ = 0.1540 nm) and scanning range 2θ = 10–70°. All diffraction patterns were acquired using a generator with a 45-kV voltage and a current of 40 mA.
2.2.2 Textural properties
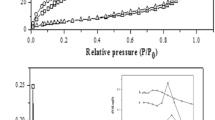
Textural properties were determined by N2 adsorption–desorption at 77 K using a Micromeritics ASAP 2000 gas sorption system. All samples were degassed at 383 K for 2 h under vacuum before analysis. Surface areas were determined by a multipoint BET method using data of the N2 adsorption isotherm and relative pressures (P/P0) ranging from 0.05 to 0.3. The pore size distributions of the different samples were evaluated by the BJH method using data of the N2 desorption isotherm.
2.2.3 Raman spectroscopy
Raman spectroscopy was performed using a Jobin Yvon Labram 300 Raman spectrometer equipped with a confocal microscope, a CCD detector, and a He–Ne laser source (λ = 633 nm). Raman spectra of the samples, used in the powder form, were recorded at ambient temperature in the 200–1400 cm−1 region with a resolution of 2 cm−1.
2.2.4 XPS
The surface compositions of the prepared samples were analyzed by X-ray photoelectron spectroscopy, using SSI-SSX-100 equipment, with a monochromatic Mg Kα radiation. The carbon (C1s) peak at 284.5 eV was used as an internal standard for the binding energies of other elements. Quantitative analysis of the peaks corresponding to sulfur, zirconium, and oxygen, in terms of elemental ratios, was carried out after the deconvolution of peaks using a Voigt function.
2.2.5 FTIR spectroscopy and adsorption of pyridine
In order to study the strength and distribution of acidic sites, the infrared spectra were recorded, using a Nicolet Magna 550 spectrometer equipped with a DTGS or MCT detector, following temperature-programmed desorption of pyridine at various temperatures ranging from 323 to 573 K. The quantification of Brönsted and Lewis acid sites was estimated from the band attributed to the vibration of protonated pyridine (Py-B) located at 1540 cm−1 and its counterpart assigned to the vibration of the coordinated pyridine (Py-L) at 1445 cm−1, using integrated molar absorption coefficients value of 1.8 and 1.5 cmμmol−1, respectively.
The previously calcinated sample (25 mg), pressed into a thin self-supported pellet, was first introduced in a specially made IR cell. The latter consists of a quartz tube glued at its end to another, relatively short Pyrex tube (to obtain a T shape), closed on both sides by CaF2 windows. The pellet was placed on a sample holder that can be moved along the tube between the sample processing and spectral recording areas. The sample was then activated in situ in a flow of O2 (30 mL/min) at 673 K for 2 h followed by outgassing at the same temperature for 1 h. After cooling to room temperature, the sample was exposed to the pyridine vapor for 30 min at 298 K. The pyridine excess was removed by outgassing under vacuum for 30 min at the desired desorption temperature until reaching a residual pressure below 10−4 Pa. Finally, FTIR spectra were recorded by performing 128 scans at a spectral resolution of 4 cm−1. It is important to note that all the spectra, shown in the present study, were obtained after subtraction of the spectra of an evacuated sample without pyridine and normalization for 25 mg of the solid.
2.2.6 Catalytic test
The n-hexane isomerization reaction was used as a model test in order to characterize the acidity of the prepared samples. Platinum was used, as reported in our previous work [29], in order to stabilize the catalytic activity and to prevent rapid deactivation caused by the coke formation. The reaction was thus performed in a continuous fixed-bed reactor, with about 100 mg of the synthesized sample mechanically mixed with a fixed amount of Pt/Al2O3 (0.35 wt% of Pt) previously reduced at 673 K, under hydrogen flow of 30 cm3.min−1. Except when otherwise stated, a (Pt/Al2O3)/(sample) weight ratio of 1 was used. Before testing, the obtained mixture was pretreated in a helium flow at 673 K for 1 h and then decreased to 523 K where it was reduced in a hydrogen flow of 20 cm3min−1 for 2 h. Finally, the temperature was decreased to the desired value, and a gas mixture of hydrogen and n-hexane was fed, at atmospheric pressure, into the reactor with a total flow of 20 cm3min−1. The temperature of liquid n-hexane was maintained at 273 K.
3 Results and discussion
3.1 Sulfur analysis
The analysis of the results presented in Table 1 show that the amount of sulfur retained by the SZP catalyst is more important compared to SZB and SZtB samples. After calcination at 833 K, the SZP catalyst contains 7.28 wt% of sulfur, while the amounts of sulfur retained by the SZB and SZtB solids were 0.30 and 1.31%, respectively. In addition, the sulfate groups developed by the SZP sample were characterized by the highest thermal stability. The percentage of sulfur, lost after heat treatment, was ca. 0.7% for this solid, but reaches 80% and 84.6% for SZB and SZtB catalysts, respectively.
3.2 Textural properties
The textural properties of different samples, treated at 833 K, are listed in Table 2. They show that all catalysts exhibit low specific surface areas and a narrow pore size distribution, with a small advantage for the SZP sample. This could be explained by the highest superficial sulfate density. In fact, it is known that oxoanions like sulfates produce a textural promotion of the catalysts and suppress the grain growth of zirconia during calcinations, leading to a relatively higher surface area. Furthermore, it was noted that the average pore diameter of the SZtB sample is bigger than those of SZP and SZB solids. This could be attributed to the large size of alcoholic groups bound to zirconium atoms in the zirconium tert-butoxide precursor which leads, after elimination of the organic species during calcination, to a slightly larger porosity.
3.3 XRD
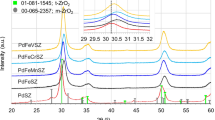
The XRD patterns of sulfated zirconia samples, after calcination at 833 K, are presented in Fig. 1. It can be seen that SZB is the best crystallized sample. In this case, zirconia exists mainly in the monoclinic phase with a low amount of metastable tetragonal phase. The ZrO2 tetragonal phase peaks are located at 2θ = 29.9° (main peak), 34.1, 34.9, 49.5, 50.2, and 59.5°, and the main peaks of the monoclinic phase are located at 28.3 and 31.6°. In the case of the SZP and SZtB samples, poor crystallization and existence of only the metastable tetragonal phase of zirconia were noted. Sulfate groups are known to suppress the zirconia grain growth during calcination and to stabilize the metastable tetragonal phase, considered as the catalytically active phase in acid-catalyzed reactions [32]. The results indicate, therefore, that the magnitude of the interactions between zirconia and sulfate species was more important for SZP and SZtB samples. The retention of sulfur by the SZP sample is much more pronounced than in the SZB and SZtB solids. As shown in Table 1, SZB catalyst was characterized by the lowest superficial sulfate density. These results are in good agreement with those reported in the literature, concerning the stabilizing effect of sulfate groups toward zirconia, and indicate that interactions between sulfate species and the zirconia surface depend upon the nature of the zirconium precursor.

XRD patterns of SZB, SZP, and SZtB catalysts
3.4 Raman spectroscopy
The Raman spectra of samples SZB and SZP calcinated at 833 K are shown in Fig. 2. The Raman spectra of the SZtB were useless due to a large fluorescence background. In the case of zirconia crystallinity, Raman spectra were consistent with XRD results. The bands in the 100–700 cm−1 region for SZB were characteristic of monoclinic zirconia [33] with intense peaks at 180, 190, 334, 348, 382, 476, 617, and 638 cm−1, whereas those obtained for SZP were broader and less intense, notably at 151, 269, 457, and 640 cm−1, which are assigned to the tetragonal phase [33], indicating a low crystallinity in this case.

Raman spectra of SZB and SZP catalysts
A very broad band with a maximum at ca. 1045 cm−1 was observed for the SZP catalyst (Fig. 2). This band is attributed to sulfate surface species [6]. A weak band was also observed for SZB at ca. 1175 cm−1 with a shoulder at 1235 cm−1 but the main band at 1045 cm−1 was not observed. This indicates lower sulfate surface species in SZB, which is confirmed below by XPS (Table 3).
3.5 FTIR spectroscopy and adsorption of pyridine
The FTIR spectra of SZP, SZB, and SZtB catalysts, collected after activation, are presented in Fig. 3. The appearance of very intense bands around 1000–1200 cm−1, due to the presence of sulfate species, was noticed except for SZtB, reflecting its much lower amount of sulfur. All samples exhibit an absorbance band close to 1400 cm−1 which can be attributed to S=O vibrations mode within SO3 molecules or pyrosulfate species, as reported for related systems by Hofmann et al [34]. Furthermore, this band was shifted to higher wave numbers in the case of the SZP sample, suggesting an important contribution of pyrosulfate species at the surface of this catalyst [35, 36].

FTIR spectra of SZP, SZB, and SZtB catalysts after activation
The FTIR spectra shown in Figs. 4 and 5 have been recorded after adsorption and desorption of pyridine molecules at various temperatures, in order to study both the type of acid sites and acidity strength of all prepared samples. Comparison of the intensities of absorption bands at 1545 and 1445 cm−1 attributed to the interactions between Brönsted and Lewis acid centers, respectively, with pyridine molecules, indicates that the SZP sample exhibits the highest density of Lewis and Brönsted acid sites. This result is consistent with textural properties, since this catalyst shows relatively the most important specific surface area. All the spectra reveal the existence of absorption bands at 1445, 1490, and 1610 cm−1 assigned to pyridine coordinated to Lewis acid sites. However, no Brönsted acid sites in the case of the SZB catalyst were observed. These results are in good agreement with those reported by Pinna et al. [37] who showed the necessity of Lewis acid sites for n-butane isomerization, and observed that catalytic activity drops to nearly zero if CO was introduced during the reaction. Yaluris et al [38] also reported the importance of Lewis acid sites in generating initial activity and that Brönsted ones are required for sustaining the catalytic activity during n-butane isomerization.

FTIR spectra of SZP, SZB, and SZtB samples after TPD of pyridine

FTIR spectra of SZP, SZB, and SZtB samples after desorption of pyridine at 423K
Figures 6 and 7 show the abundance of Lewis and Brönsted acid sites after desorption of pyridine at various temperatures. First, a decrease of the amount of both Lewis and Brönsted acid sites, with increasing desorption temperature, was noted. Second, in the case of SZP and SZtB samples, Lewis acid sites were slightly more abundant than those of Brönsted type. However, the persistence of Lewis and Brönsted acid sites, even after pyridine desorption at 573 K, shows that these acid sites are relatively strong. Furthermore, it appears that Brönsted acid sites, developed by the SZtB sample, reveal a better resistance, when the pyridine desorption temperature was increased, than those existing on the surface of the SZP catalyst. This result indicates that SZtB catalyst exhibits stronger Brönsted acid sites.

Lewis acidity measured by TPD of adsorbed pyridine of SZP, SZB, and SZtB samples

Brönsted acidity measured by TPD of adsorbed pyridine of SZP, SZB, and SZtB samples
3.6 XPS
The results of XPS analysis are shown in Table 3. The O1s, S2p, and Zr3d5 binding energies were consistent with those reported in literature [39, 40] for such catalysts. No influence of zirconium precursor on the binding energies could be detected. The solids differ by their S/Zr superficial ratio. Indeed, we note that the SZP sample shows the highest ratio. However, it is important to note that in this case, the majority of the sulfate groups seem to be trapped in the bulk, since the results summarized in Table 1 show a much higher sulfur content in the case of solid SZP.
The highest S/Zr ratio in the case of the SZP sample may indicate the existence of polysulfate species on the surface of this solid. This might be confirmed by the results of XRD studies and Raman and FTIR spectroscopies which showed, on the one hand a delay in crystallization, and in the crystallographic phase transition of zirconia, and on the other hand the existence of a broad band at ca.1045 cm−1 attributed to surface sulfate species.
3.7 Catalytic activity
3.7.1 Catalytic properties
The results obtained during n-hexane isomerization reaction tests at atmospheric pressure are shown in Figs. 8 and 9. The SZP catalyst exhibits a very high initial activity compared to SZB and SZtB samples. However, the initial selectivity toward isomerization products was low for SZP (27.3%). All catalysts, especially the SZP sample, deactivated rapidly before reaching a stationary state after almost 2 h onstream. Then, the overall activity and selectivity to isomerization products were almost similar for all solids.

Conversion of n-hexane at 473 K using SZB, SZP, and SZtB catalysts and a weight ratio of ((Pt/Al2O3)/sample) = 1

Selectivity toward isomerization during n-hexane isomerization reaction at 473 K using SZB, SZP, or SZtB catalysts and a weight ratio of ((Pt/Al2O3)/sample) = 1
Figure 10 shows the initial conversion during n-hexane isomerization reaction and the superficial density of Lewis acid obtained using the FTIR of adsorbed pyridine results. It appears that there is a pronounced dependence of these two parameters. In fact, the catalytic activity was higher for the SZP sample which also exhibits a higher density of Lewis acid sites, whereas the solid, showing the smallest density of Lewis acid sites (SZtB), presents the lowest catalytic performances. Furthermore, we note that the SZB catalyst exhibits a relatively important initial activity, in spite of the absence of Brönsted acid sites, as indicated by FT-IR of adsorbed pyridine (Fig. 7). This is in further agreement with the results reported by Pinna et al [37] and Yaluris et al [38], indicating the importance of the presence of Lewis acid sites in n-alkane isomerization reactions.

Variation of the initial conversion at 473 K with the amount of Lewis acid sites for SZB, SZP, and SZtB catalysts
3.7.2 Influence of platinum loading on catalytic performances
The very high initial conversion, accompanied by a rapid deactivation, observed for the SZP catalyst, could be explained by the presence of very strong acid sites on the surface of this sample. These acid sites are able to form rapidly carbenium species during the isomerization process, which were advocated as coke precursors. For instance, when carbenium ions are generated, in the absence of preformed active hydrogen atoms, capable of transforming them into isomers, these carbenium species will polymerize and form coke. This may indicate that a high density of Pt particles is required in order to increase the active hydrogen concentration.
In order to overcome the problem of rapid deactivation of the SZP catalyst, and to improve the catalytic activity as well as the stability of this catalyst during n-hexane isomerization reaction, the determination of the optimal quantity of Pt/Al2O3 needed to preserve the catalytic activity of the SZP sample as high as in the initial state, was studied.
Figure 11 exhibits the variation of the conversion toward isomerization with the (Pt/Al2O3)/(SZP) weight ratio. It appears first that the initial conversion of n-hexane increases considerably when the (Pt/Al2O3)/(SZP) weight ratio was varied from 0.5 to 1. However, we note a small improvement of catalytic performances when the (Pt/Al2O3)/(SZP) weight ratio was higher than 1. Table 4 shows the variation of the amount of sulfur lost during the n-hexane isomerization reaction by the SZP sample as a function of (Pt/Al2O3)/(SZP) weight ratio. This shows that sulfur loss increases considerably when the amount of the used Pt increases. These results suggest that the observed fragility of sulfate species is due to the presence of platinum particles.

Variation of n-hexane conversion at 443 K toward isomerization products using different (Pt/Al2O3)/SZP weight ratios
We investigated the products emitted during TPR–H2 experiments, carried out on a mechanical mixture of the SZP sample and Pt supported on alumina with a weight ratio equal to 1, in order to confirm the relationship between the presence of Pt particles and the loss of sulfur during the isomerization reaction which is directly related to the decrease of the catalytic activity. The emitted gases were identified by mass spectrometry. For this purpose, two TPR–H2 experiments have been carried out. In the first one, the mechanical mixture of Pt/Al2O3 with the SZP solid was not pretreated under H2, while in the second, the mechanical mixture was pretreated under H2 at 673 K during 1 h. The results obtained are presented in Figs. 12 and 13, respectively.

Desorbed gas during TPR–H2 experiments using a 1:1 mixture of SZP and 1 wt% Pt/Al2O3 without pretreatment under H2

Desorbed gas during TPR–H2 experiments using a 1:1 mixture of SZP and 1 wt% Pt/Al2O3 pre-treated under H2 at 673K
The mixture which did not undergo a pretreatment under H2 atmosphere showed the existence of two broad peaks, assigned to H2O, produced during the reduction process. The first is situated between 343 and 473 K and could be attributed to the physisorbed water molecules, whereas the second peak in the range 703–773 K, corresponds to the dihydroxylation of the sample surface. Moreover, two intense peaks at 688 and 723 K were observed, indicating the loss of sulfur retained by the SZP sample as SO2 and H2S gases, respectively.
However, in the case of the TPR experiment using a mixture which already underwent a pretreatment under H2 at 673 K during 1 h, we noticed the disappearance of the two peaks related to the emission of SO2 and H2S gases. These results suggest that the initial retained sulfur has been already eliminated during the pretreatment step and confirm that the presence of the platinum particles facilitated the sulfur loss during the n-hexane isomerization reaction in the presence of H2.
4 Conclusion
Sulfated zirconia catalysts were prepared using the sol–gel process. Hydrolysis water was produced in situ. The effects of the zirconium alkoxide on the physicochemical properties and the acidity of the samples have been investigated. The results show that the interaction between zirconia and sulfate species is more pronounced when zirconium propoxide was used as a precursor. In this case, the highest superficial sulfate density with a large band at 1050 cm−1 detected by Raman spectroscopy was observed. This could be attributed to the existence of the polysulfate groups. In addition, this sample shows a stabilization of the metastable tetragonal phase of ZrO2 at 833 K. It was also found that all prepared samples exhibit low specific surface areas and narrow pore distributions. The characterization of the acidity of different samples, by FTIR of adsorbed pyridine, indicated the highest density of Lewis and Brönsted acid sites in the case of the SZP catalyst. However, no Brönsted acid sites were present in the case of the SZB sample. The catalytic results showed that the SZP catalyst exhibits a very high initial conversion during the n-hexane isomerization reaction. However, the selectivity toward the isomerization products was low and this catalyst deactivated rapidly.
The effects of the platinum amount on the stability of sulfated zirconia catalysts were also studied, and showed a slightly higher selectivity toward isomerization, when the amount of Pt increases. In this case, however, sulfur loss as H2S and SO2 species also increased considerably, leading to a rapid deactivation of the catalyst.
References
Grau JM, Yori JC, Parera JM (2001) Appl Catal A 213:247–257
Yori JC, Gastaldo RJ, Benitez VM, Pieck CL, Vera CR, Grau JM (2008) Catal Today 133-135:339–343
Hino M, Kobayashi S, Arata K (1979) J Am Chem Soc 101:6439–6441
Song X, Sayari A (1996) Catal Rev Sci Eng 38:329–412
Zhang S, Zhang Y, Tierney JW, Wender I (2000) Appl Catal A 193:155–171
Reddy BM, Patil MK (2009) Chem Rev 109:2185–2208
Reddy BM, Sreekanth PM, Reddy VR, (2005) J Mol Catal 225:71–78
Cui X, Ma HZ, Wang B, Chen HW (2007) J Hazard Mater 147:800–805
Das SK, Bhunia MK, Sinha AK, Bhaumik A (2009) J Phys Chem C 113:8918–8923
Wang S, Matsumura S, Toshima K (2007) Tetrahedron Lett 48:6449–6452
Salomatina OV, Kuznetsova TG, Korchagina DV, Paukshtis EA, Moroz EM, Volcho KP, Barkhash VA, Salakhutdinov NF (2007) J Mol Catal A 269:72–80
Lutecki M, Breitkopf C (2009) Appl Catal A: Gen 352:171–178
Calbro DC, Vartuli JC, Santiesteban JG (2002) Top Catal 18:231–242
Föttinger K, Halwax E, Vinek H (2006) Appl Catal A: Gen 301:115–122
Manoilova OV, Olindo R, Arean CO, Lercher JA (2007) Catal Commun 8:865–870
Busto M, Shimizu K, Vera CR, Grau JM, Pieck CL, M.A. D’amato, Causa MT, Tovar M (2008) Appl Catal A 348:173–182
Yadav GD, Nair JJ (1999) Micro Mesopor Mater 33:1–48
Banerjee B, Bhunia S, Bhaumik A (2015) Appl Catal A 502:380–387
Farcasciu D, Li JQ, Cameron S (1997) Appl Catal A 154:173–183
Bokhimi X, Morales A, Novaro O, Lopez T, Gomez R (2000) Polyhedron 19:2283–2287
Föttinger K, Zorn K, Vinek H (2005) Appl Catal A 284:69–75
Signoretto M, Oliva L, Pinna F, Strukul G (2001) J Non-Cryst Solids 290:145–152
Lutecki M, Breitkopf C (2009) Appl Catal A 352:171–178
Belido AF, Klabunde KJ (1998) J Catal 176:448–458
Ward DA, Ko EI (1995) J Catal 157:321–333
Armendariz H, Coq B, Tichit D, Dutartre R, Figuéras F (1998) J Catal 173:345–354
Morterra C, Cerrato G, Di Ciero S, Signoretto M, Pinna F, Strukul G (1997) J Catal 165:172–183
Ben Hamouda L, Ghorbel A (2000) J Sol-GelSci Technol 19:413–416
Hamouda LB, Ghorbel A (2006) J Sol-Gel Sci Technol 39:123–130
Ward DA, Ko EI (1994) J Catal 150:18–33
Signoretto M, Pinna F, Strukul G, Chies P, Cerrato G, Di Ciero S, Morterra C (1997) J Catal 167:522–532
Stichert W, Schüth F, Kuba S, Knözinger H (2001) J Catal 198:277–285
Xie S, Iglesia E, Bell AT (2000) Chem Mater 12:2442–2447
Hofmann A, Sauer J (2004) J Phys Chem B 108:14652–14662
Bensitel M, Saur O, Lavalley JC, Morrow BA (1988) Mater Chem Phys 19:147–156
Föttinger K, Zorn K, Vinek H (2005) Appl Catal A 284:69–75
Pinna F, Signoretto M, Strukul O, Cerrato G, Morterra C (1994) Catal Lett 26:339–344
Yaluris G, Larson RB, Kobe JM, Gonzaılez MR, Fogash KB, Dumesic JA (1996) J Catal 158:336–342
Breitkopf C, Matysik S, Papp H (2006) Appl Catal A 301:1–8
Coman S, Pârvulescu V, Grange P, Pârvulescu VI (1999) Appl Catal A 176:45–62
Acknowledgements
The authors thank Prof. M. Houalla and Dr. G. Clet (ENSICAEN, France) for help and assistance in Raman measurements.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ben Hammouda, L., Ghorbel, A. Influence of the zirconium precursor on the acidic and catalytic properties of sulfated zirconia catalysts prepared by sol–gel process. J Sol-Gel Sci Technol 89, 543–552 (2019). https://doi.org/10.1007/s10971-018-4877-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-018-4877-6