
Abstract
In this study, preparation of Sn doped (0–30 mol % Sn) TiO2 dip-coated thin films on glazed porcelain substrates via sol–gel process have been investigated. The effects of Sn content on the structural, optical, and photo-catalytic properties of applied thin films have been studied by X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy (SEM), field emission SEM (FE-SEM), and high resolution transmission electron microscopy (HR-TEM). Surface topography and surface chemical state of thin films were examined by atomic force microscope (AFM) and X-ray photoelectron spectroscopy (XPS). XRD patterns showed an increase in peak intensities of the rutile crystalline phase by increasing the Sn dopant. The prepared Sn-doped TiO2 photo-catalyst films showed optical absorption edge in the visible light area and exhibited excellent photo-catalytic ability for degradation of methylene blue solution under UV irradiation. The result shows that doping an appropriate amount of Sn can effectively improve the photo-catalytic activity of TiO2 thin films, and the optimum dopant amount is found to be 15 mol%. The Sn4+ dopants substituted Ti4+ in the lattice of TiO2 and increased surface oxygen vacancies and the surface hydroxyl groups. TEM results showed small increase in planar spacing (was detected by HR-TEM caused by Sn dopants in titania based crystals).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The titanium dioxide has been widely used in the field of pollutant degradation and environment protection since photo-catalytic function of titania was discovered in 1972 [1, 2]. The titanium dioxide has the advantage of not only high photo-catalytic activity, but also good acid resistance, low cost, and no toxicity, which makes the titanium dioxide become one of the best photo-catalytic agents [3, 4].
TiO2 can catalytically decompose a large number of organic and inorganic pollutants under illumination of UV light [5–7]. However, depending on the structural form, the photo-catalytic activity of TiO2 has been found to vary.
Recently, most of the investigations have been focused on preparing TiO2 catalysts that can be activated by Vis-light because there is much more energy produced by the sunlight in the visible lights regime compared to the UV spectrum [8–10]. For this purpose, doping or combining TiO2 with various metals (Au, Pt, Ir) or non-metal ions has been considered [11, 12]. The photo-catalytic activity of TiO2 has been found to vary with its structural form and is reportedly higher in the anatase form compared to the rutile phase structure [13].
Lin et al. [14] and Maruska and Ghosh [15] have related the higher photoactivity of anatase TiO2 (E g = 3.2 eV) to its higher Fermi level compared to that of rutile TiO2 (E g = 3.0 eV). However, high photo-catalytic activity has been reported with multi-phased mesoporous TiO2, consisting of anatase and rutile [16, 17]. Thus, more investigations seem necessary to understand the effect of metal ion doping on the structural and optical properties of TiO2.
There are some studies on metal ion doping of TiO2 and it has been found that the anatase to rutile phase transformation is affected to a varying extent, and so is its photo-catalytic activity. The choice of the doping type may play a crucial role in the crystalline structure and the stable phase of TiO2 thin films. The metal ions which are found to inhibit the anatase to rutile phase transformation are Si [18], W, Nb, Ta, Cr [19], while the metal ions which are reported to promote the phase transformation are Ni, Co, Mn, Fe, Cu [20], V [19] and Ag [21].
Few papers are dedicated to the investigation of Sn doped titania films as photo-catalysts [9, 14, 22]. Moreover, there is a discrepancy in the literature about the crystalline stability of TiO2–SnO2 system. It is claimed that the SnO2 in TiO2–SnO2 binary oxides stabilizes the anatase phase [23, 24]. In contrast, other authors have proved that the Sn promotes the phase transformation from anatase to rutile phase structure [25, 26].
In the present work, we are discussing preparation of a novel aqueous and economically beneficial method. Here Sn has been selected as an additive to increase the photo-catalytic activity of thin films at the visible light wave lengths. The thin films were applied by sol–gel dip-coating process. Developing more stable rutile phase TiO2 thin films at low temperature was achieved in this study.
2 Experimental procedures
2.1 Preparation of the thin films
The preparation of precursor solution for Sn-doped TiO2 thin films is described as follows: TiO2 and SnO2 sols were prepared separately. For the preparation of TiO2 sol, titanium (IV) butoxide (TBT = Ti (OC4H9)4, Aldrich) was selected as titanium source. First, 0.35 mol ethanol (EtOH, Merck) and 0.04 mol ethyl acetoacetate (EAcAc is as a sol stabilizer during preparation of sol and also chelating agent during coating, Merck,) were mixed, and then 0.01 mol TBT was added by the rate of 1 mL/min to the mixture at the ambient temperature (25 °C). The solution was continuously stirred for 45 min, followed by the dropping of HNO3 as catalyst to the solution until pH = 3. De-ionized water was added to the solution slowly to initiate hydrolysis process. Solution was aged for 24 h in order to complete all reactions. The chemical composition of the resultant alkoxide solution was TBT:H2O:HNO3:EAcAc:EtOH = 1:10:1:4:35 in molar ratio. In order to prepare SnO2 sol, stannous chloride (SnCl2·2H2O, Merck) was dissolved in EtOH with molar ratio of SnCl2·2H2O:EtOH = 1:35 at ambient temperature with continuous stirring. Solution was aged for 24 h in order to complete all reactions. Then, mixtures of TiO2 and SnO2 sols were made with different mol ratios of SnO2 at the ambient temperature. The mol ratios of Sn in mixtures were 0, 1, 5, 10, 15, 20, 25, 30 mol %. The Sn-doped thin films were applied on the glazed porcelain substrates with the prepared sol solutions by dipping-withdrawing at room temperature with withdrawal speed of 10 mm/min.
The porcelain composition was Al2O3: 45.12, SiO2: 43.83, BaO: 6.18, K2O: 3.63, Na2O: 0.58, MgO: 0.39, CaO: 0.27 weight percent. Glazed composition contained SiO2: 64.45, Al2O3: 16.56, CaO: 6.07, K2O: 5.1 MgO: 4.36, Fe2O3: 3.46 weight percent. Porcelain and glaze compositions were reported by substrate manufacturer with formal report.
Substrate dimensions were 30 × 15 × 1 mm. The substrate were degreased in ethanol and cleaned in de-ionized water for 15 min and acetone with supersonic wave. The treated substrate samples were dried at 80 °C for 1 h. In order to prevent diffusion of porcelain substrate and glaze elements to final thin film, one interlayer (buffer layer) SiO2 thin film with withdrawal speed of 10 mm/min was applied. Silica sol contains tetra ethyl orthosilicate (TEOS, Merck): EtOH:H2O:HCl with 1:3.8:1:0.01 molar ratios and calcined at 400 °C for 1 h. Samples were dried at 100 °C for 1 h after each coating cycle and finally heated at a rate of 2 °C/min and calcined at 475 °C for 1 h in air atmosphere.
Substrates were coated with one SiO2 (buffer layer) and three Sn-doped TiO2 layers.
2.2 Characterization methods
The sol particle size distribution and viscosity were determined by dynamic light scattering technique (DLS-Horiba-LB 550). Differential thermal analysis (DTA) and thermogravimetric (TG) were used in order to determine crystallization temperature with a heating rate of 10 °C/min (STA 1460 equipment). XRD pattern and phase identification of thin films were recorded using X-ray diffraction analysis (Philips, MPD-XPERT, λ: Cukα = 0.154 nm). The samples were scanned in the 2θ range of 20°–70°. Raman spectra were conducted on a Renishaw RM1000 spectroscope with laser light wavelength of 514 nm. Morphology of the thin films was observed using scanning electron microscopy (SEM, XL30 Series) with an accelerating voltage of 10–15 kV. Also surface topography of the thin films was investigated using non-contact mode atomic force microscopy (AFM, Solver P47H Instrument). X-Ray photoelectron spectroscopy (XPS) spectra were recorded by a PHI 5000C ESCA spectrometer using Mg Ka radiation (hυ = 1253.6 eV). The pressure of the analyzer chamber was maintained at 5 × 10−8 Pa. Spectra were calibrated with respect to the C1s peak at 284.6 eV.
In order to make TEM foils, the films were removed from the substrates by water flotation. Droplets of the sol–gel material were also deposited on plain (un-coated) copper grids so as to form free-standing films for in situ hot-stage experiments in the TEM. Conventional TEM imaging and electron diffraction analysis was carried out using an EM-2100F-JEOL-Tokyo-Japan with acceleration voltage of 200 kV.
2.3 Photo-catalytic activity measurement
The photo-catalytic activity was evaluated by monitoring the degradation of MB solution (5 ppm) under UV illumination. The samples were placed in 20 mL aqueous MB solution and were kept in dark for 2 h. They were then irradiated from top using a UV lamp (Philips 8 W, 360 nm). The intensity of the MB characteristic band at 660 nm (I 660) in the obtained UV–vis spectrum (Varian cary 50 spectrophotometer) was used to determine the concentration of MB in the solution (C t ).
The degradation rate of MB, which represents the photo-catalytic efficiency of the films, can be determined by Eq. 1.
where d is degradation rate, C t is concentration after radiation and C 0 is concentration before radiation [27].
3 Results and discussion
Homogeneous and transparent sols were obtained for all compositions. Viscosity of all sols was measured at 25 °C. In all samples, a Newtonian behavior was observed. Physical properties of sols are presented in Table 1. As seen, all sols revealed a good stability for more than 25 days. Particle size distribution and mean particle size (d 50) of sols decrease by increasing SnO2 content. Also, slight increase was observed in viscosity of sols.
TiO2 sol was heated up to 100 °C for 1 h for gel formation. TG-DTA curve of TiO2 gel (titania based precursor gel) from ambient temperature up to 700 °C is shown in Fig. 1. The sample lost weight gradually with the increase of temperature until about 600 °C. After 600 °C the change in weight was small. The total weight loss was 27.7% with respect to the original sample weight.

DTA curve of the gel (titania based precursor gel) from ambient temperature up to 700 °C (Heating rate = 10 °C/min)
Also, it shows an endothermic peak at 92 °C (50–100 °C) which is attributed to evaporation of absorbed water and solvent (EtOH) of the gel. The first exothermic peak at 129 °C (120–140 °C) is probably attributed to combustion of residual organic component of TBT precursor. It is clear that titania precursor gel is thermally stable from 140 °C up to 280 °C. Small exothermic peak at temperature of 298 °C (290–340 °C) is probably corresponds to combustion of EAcAc (auto ignition temp. = 295 °C).
There are also two exothermic peaks, first at about 400–470 °C corresponds to the crystallization of anatase phase of TiO2 and the other at 495 °C (460–520) corresponds to the phase transformation of anatase to rutile of titania [28, 29].
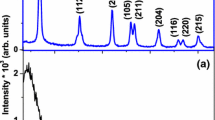
Figure 2 shows the XRD patterns of Sn-doped TiO2 thin films with different mol ratios of Sn heat treated at 475 °C for 1 h. X-Ray diffraction peak at 25.5° corresponds to characteristic peak of crystal plane (1 0 1) of anatase, at 27.6° corresponds to characteristic peak of crystal plane (1 1 0) of rutile in films. According to the XRD patterns, the pure TiO2 (T) constituted of pure anatase phase. No characteristic peaks of SnO2 were observed in Sn-doped TiO2 thin films up to 25% Sn (T-25%S), which suggests the incorporation of Sn4+ into the TiO2 lattice [30]. As seen, T-30% Sn was consisted of high percentage of SnO2 phases. So, we deleted this composition from this study. The diffraction peaks of rutile crystallites appeared in X-ray profiles with increasing Sn dopant in thin films. The addition of Sn has a promoting effect on the transformation of anatase to rutile crystalline phase [31, 32], due to the rutile like structure of SnO2-cassiterite phase. It is also found that the peak position of (1 0 1) anatase and (1 1 0) rutile planes shift to a smaller diffraction angle with increasing Sn content in TiO2 thin films. This may results from the exchange of Ti with Sn in the TiO2 lattice.

XRD patterns of dip-coated pure (T) and Sn-doped TiO2 thin films (T-x mol% Sn) with different Sn mol%, calcined at 475 °C on porcelain substrates (A Anatase, R Rutile, S SnO2)
Also, Since the electronegativity and ionic radius of Sn4+ ion (1.8, 0.71°A) approach those of Ti4+ ion (1.5, 0.6°A) in TiO2 [33] it is expected that the Sn4+ ions will replace lattice Ti4+ ions and thus occupy lattice Ti4+ positions in the doping reactive process. The ionic radius of the doping Sn4+ ion is larger than that of the lattice Ti4+ ion. This will induce a distortion of the lattice and increment the lattice parameters and cell volume of Sn-doped TiO2 thin films compared with those of pure TiO2.
The average crystallite size of thin films (D) was determined from the XRD patterns, according to the Scherrer equation (2) [34]
where k is a constant (shape factor, about 0.9), λ the X-ray wavelength (0.154 nm), β the full width at half maximum (FWHM) of the diffraction peak and θ is the diffraction angle. The values of β and θ of anatase and rutile phases were taken from anatase (1 0 1) and rutile (1 1 0) planes diffraction lines, respectively. The amount of rutile in the samples was calculated using the following equation [35]
where X R is the mass fraction of rutile in the samples, and I A and I R are the X-ray integrated intensities of (1 0 1) reflection of the anatase and (1 1 0) reflection of rutile, respectively. The diffraction peaks of crystal planes (101), (200) and (105) of Anatase phase in XRD patterns were selected to determine the lattice parameters of the TiO2 and Sn-doped TiO2 thin films. The lattice parameters were obtained by using the Eq. 4 [34]
where d (hkl) is the distance between the crystal planes of (hkl); λ is the wavelength of X-ray used in the experiment; θ is the diffraction angle of the crystal plane (hkl); hkl is the crystal plane index; and a, b, and c are lattice parameters (in Anatase form, a = b ≠ c).
The characteristic of Sn-doped TiO2 thin films after thermal treatment at 475 °C is reported at Table 2. By increasing Sn dopant, transformation of anatase to rutile phase was promoted, the amount of rutile phase structure was increased, and a gradual decrease in the crystalline size was detected. The decrease in crystal size can be attributed to the presence of Sn–O–Ti in the Sn-doped TiO2 thin films which inhibits the growth of crystal grains [36].
Also, the lattice parameters and cell volume of the TiO2 and Sn-doped TiO2 thin films are summarized in Table 2. It is obvious that the lattice parameters and cell volume of the Sn-doped TiO2 thin films increase with increasing amount of Sn4+ substitution for Ti4+ in rutile lattice, up to the nominal 25%, meaning that rutile structure is saturated with Sn. In T-30%S, there are so many Sn ions to enter TiO2 lattice, and some Sn atoms are present as SnO2 phase as shown in Fig. 2. In T-15%S sample a significant change in tetragonality of titania crystal structure was seen. This change contains extreme decrease in c-axis length and cell volume.
Raman spectroscopy was also applied as an additional characterization method to study the phase transformation of titania at presence of Sn dopant. The anatase and the rutile phase have different Raman active modes. The anatase main peaks are at 395 (B 1g), 513 (A 1g) and 639 cm−1 (E g) and the rutile peaks are at 447 and 610 cm−1 [37].
Figure 3 indicates the Raman spectra of the undoped and Sn-doped TiO2 thin films with different Sn dopant calcined at 475 °C. The Raman spectra of the films doped with different Sn dopant showed broadening with increasing Sn content.

Raman spectra of Sn-doped TiO2 thin films (T-x mol% S) for x = 0, 1, 5, 10, 15, 20 and 25 mol% calcined at 475 °C
In doped samples, mixtures of anatase and rutile phase were presented. This result is already presented by XRD studies and is confirmed by the Raman spectroscopy. Sn did not cause appearance of any additional bands but increasing Sn dopant has other obvious effects on the frequencies of the fundamental vibrations, like showing a general shift of vibrational modes to lower frequencies with increasing Sn dopant.
The surface composition and chemical states of Sn-doped TiO2 thin films studied by XPS method, are shown in Fig. 4. It shows the XPS spectra of Ti 2p (a), Sn 3d (b) and O 1s (c and d). The binding energy of Ti 2p3/2 and Ti 2p1/2 were observed at approximately 458.5 and 464.6 eV, respectively, indicating that the Ti element mainly existed as the chemical state of Ti4+ [38]. Interestingly, the binding energies of Ti 2p3/2 and Ti 2p1/2 shifted to lower values after the addition of dopant Sn4+ compared to pure TiO2 (Table 3; Fig. 4a). This shift can prove the existence of Ti with lower valence. The formation of Ti with lower valence can be attributed to the existence of oxygen vacancies in the thin film after doping Sn.

a Ti 2p, b Sn 3d, c O 1s XPS core level spectra of Sn-doped TiO2 samples and d O 1s fitting results for sample T-15%S
XPS spectra of Sn 3d region are given in Fig. 4b. Two peaks corresponding to Sn 3d5/2 and Sn 3d3/2 are observed. The peak position corresponding to Sn 3d5/2 (Table 3) is located between those of Sn 3d5/2 in SnO2 (486.5 eV) and Sn 3d5/2 in metallic Sn (485.0 eV), which ascribed to incorporated Sn4+ in the lattice of TiO2 [8].
The XPS spectra of O 1s core level of T-x mol%S samples are asymmetric (Fig. 4c), indicating that there are at least two kinds of chemical states. Close to the peak corresponding to oxygen in TiO2 lattice (OL), the other peak at higher binding energy can be identified (Fig. 4d) as Ti–OH resulting from the chemisorbed water (OH) [38, 39]. The results listed in Table 3, indicate further XPS data. As seen, the amount of surface hydroxyl oxygen and oxygen vacancies are enhanced by doping Sn4+. The oxygen vacancies are active groups, which can easily combine with other atoms or groups and become stable. It accounts for an increase in the amount of surface hydroxyl oxygen. Increase in surface hydroxyl is favorable for photo-catalytic reactions [1]. The hydroxyl content (OL and OH %) is the ratio of the area of corresponding peak to the total area of the two O 1s peaks (Table 3).
Figure 5 shows the results of photo-catalytic decomposition of MB solution caused by degradation of MB in contact with thin films with different Sn content. The degradation amount of MB increased by increasing Sn dopant in thin films up to 15 mol% and then decreased at higher Sn dopant. The maximum degradation is about 73.4% for T-15%S sample. The photo-catalytic activities of all the doped Thin films are higher than the pure TiO2 film (except 25% Sn).

Photo-catalytic decomposition profile of MB. Over different Sn-doped TiO2 nanocrystalline thin film calcined at 475 °C for 1 h
This phenomenon is attributed to two reasons stated below. First, it is due to the increase of surface hydroxyl of doped thin films [39]. Second, it is because of decrease in recombination rate of photo-induced carriers in Sn-doped thin films. The conduction band energy levels for SnO2 and for TiO2 are different, 0 eV for SnO2 (vs. NHE, pH = 7) and −0.5 eV for TiO2 (vs. NHE, pH = 7) [40]. Due to this potential difference, the photo-induced electrons transfer from the surface of TiO2 to that of SnO2 can take place easily when Sn is doped into TiO2. On the other hand, however, if there is too much doped Sn, the relative density of TiO2 decreases which cause the activity to decrease. In our case it is around 15 mol% of Sn that makes the photo-catalytic activity change from increasing to decreasing.
Also the Fermi levels of SnO2 is lower than TiO2, which leads to the photo-induced electrons transfer from the conductive band of TiO2 to that of SnO2 on the surface of TiO2, meanwhile, the raised Fermi level makes the driven force of the photo-induced electrons from TiO2 to SnO2 increase, these factors can also increase the separation ratio of photo-induced electrons and holes.
Figure 6 shows the UV–vis spectra of the pure TiO2 and the Sn-doped TiO2 thin films deposited on glazed porcelain substrates in the wavelength range of 250–600 nm. In this figure, the absorption edge of Sn-doped TiO2 thin films shifts towards longer wavelengths with increasing Sn dopant content. It indicates a decrease in the energy band gap (inset in Fig. 6). It has been reported that metal doping could form a dopant energy level within the band gap of TiO2 [8, 41, 42]. A dopant of Sn4+ has also been found to be located approaching and below the conduction band [8]. Therefore, the red shift of the absorption edge for the Sn-doped TiO2 should be the result of the electronic transition from the valance band to the doping energy level. On the other hand, according to the XRD result, doping of the Sn accelerated the transformation from anatase to rutile phase of TiO2, and the rutile phase has a band gap smaller than that of anatase, so this transformation should be another reason for the red shift.

UV–vis absorption spectra of pure TiO2 and Sn-doped thin films deposited on glazed porcelain substrate (four coating cycle times, calcined at 475 °C) with calculated energy band gaps of samples
The reported optical band gap (E g) in Fig. 6 (inset) has been calculated using the UV–vis spectra by:
where hν is the photon energy, A and n are constants. For allowed direct transition n = 1/2, direct forbidden transition n = 3/2 and indirect allowed transition n = 2 [43]. The optical band gap Energy (E g) is found by extrapolating the straight line portion of (αhν)1/2 with the abscissa axis (hν) in the vicinity of the fundamental optical transition for pure and doped thin films. For Sn dopant more than 5%, because of the presence of high percent of rutile phase (shown in Table 2), the band gaps are similar and lower than rutile phase. These results show that doped thin films have a higher absorbance at visible light. Also, it was found that the E g shows a decrease as the Sn dopant in the films increased. This behavior is similar to TiO2 films with implanted tin ions [44].
SEM micrographs of T-15%S thin film, with improved photo-catalytic activity, are shown in Fig. 7. Thin film showed intergranular spaces and discontinuities with slightly defined grains at different magnifications [Fig. 7a (lower mag.), b (high mag.)]. By increasing Sn dopant content, surface displays a low porous structure with a particle size around 100 nm (not shown in this figure). This figure also shows formation of crack-free thin film on glazed porcelain substrate.

a, b SEM micrographs of T-15%S at different magnification and c cross-section image of the thin film calcined at 475 °C for 1 h
Figure 7c shows the cross-section image of the prepared coating capture by FE-SEM. As the figure shows, the thickness of thin film with 15 mol % Sn is about 150 nm. Thin film structure shows a parallel columnar like structure.
Figure 8 shows AFM topography (3D) and surface roughness of T-15%S sample. It can be seen that thin film has a columnar-like morphology (Fig. 8a) with a root mean square (rms) roughness of 14.5 nm with a distribution of grain size of about 50–70 nm (Fig. 8b). AFM images indicate that the films are not continuous showing some surface porosity, which make them adequate for photo-catalytic applications.

a AFM profile image (1 μm × 1 μm) of T-15%S and b surface roughness of the thin film calcined at 475 °C for 1 h
Figure 9 shows the cross-section TEM images of undoped TiO2 (Fig. 9a) and surface of T-15%S (Fig. 9c). The electron diffraction (ED) patterns of the selected area on pure TiO2 sample (inset of Fig. 9a) shows the strong Debye–Scherrer rings indicating the anatase phase structure crystallite. Figure 9c shows the strong Debye–Scherrer rings and complicated bright spots, indicating the coexistence of polycrystalline anatase and rutile crystallites. This agrees with the aforementioned conclusion that the Sn4+ ions doped in TiO2 can promote the formation of rutile phase structure titania.

TEM and HRTEM image of undoped TiO2 thin film (a, b) and T-15%S (c, d)
Figure 9b, d displays the HRTEM images of undoped and doped TiO2 thin film, respectively. For anatase structure, the fringe spacing (d) of (101) crystallographic plane is determined to be 3.50 Å for undoped TiO2 film (Fig. 9b) and 3.52 Å for T-15%S (Fig. 9d). This implies that Sn4+ ions are doped into TiO2 lattice in substitutional space in T-15%S, since the ionic radius of Sn4+ (0.71°A) is larger than that of the lattice Ti4+ (0.6°A), a fringe spacing of 3.35 Å corresponding to the (110) planes of rutile TiO2 is observed in T-15%S (Fig. 9d). All of these are consistent with the aforementioned XRD results.
4 Conclusion
In this research, Sn-doped TiO2 thin films with photo-catalytic properties have been prepared by a sol–gel dip-coating method. The thickness of applied thin films on porcelain substrates was about 150 nm. The photo-catalytic activity of the doped thin film is higher than that of pure TiO2 thin films. Sn4+ substitution for Ti4+ in the TiO2 lattice results in a decrease in the rate of photogenerated electron–hole recombination that is responsible for the enhancement in photo-catalytic degradation rate.
The main results of the present study can be summarized as follows: (1) the band gap of Sn-doped TiO2 thin films vary continuously from those of pure TiO2 to those of pure SnO2 with increasing Sn dopant, and (2) Sn substitution for Ti in TiO2 rutile lattice structure increased the oxidation–reduction potential of the oxide and showed the lowest indirect energy transition. These effects are believed to inhibit the photogenerated electron–hole recombination, and thus endow the enhanced photoactivity for TiO2-x mol% Sn thin film.
References
Fujishima A, Honda K (1972) Nature 238:37–38
Samuneva B, Kozhukharov V (1993) Mater Sci 28:2353–2360
Wang CY, Liu CY, Shen T (1997) J Photochem Photobiol A Chem 109:65–70
Palmer FL, Eggins BR (2002) J Photochem Photobiol 148:137–143
Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Chem Rev 95:69–96
Ohko Y, Fujishima A, Hashimoto K (1998) J Phys Chem B 102:1724–1729
Hermann JM (1999) Catal Today 53:115–129
Cao Y, Yang W, Zhang W, Liu G, Yue P (2004) New J Chem 28:218–222
Liu Q, Wu X, Wang B, Liu Q (2002) Mater Res Bull 37:2255–2262
Fujishima A, Rao TN, Tryk DA (2000) J Photochem Photobiol C Photochem Rev 1:1–21
Subramanian V, Wolf E, Kamat PV (2001) J Phys Chem B 105:11439–11446
Rajeshwar K, Tacconi NR, Chenthamarakshan CR (2001) Chem Mater 13:2765–2782
Tsai SJ, Cheng S (1997) Catal Today 33:227–237
Lin J, Yu JC, Lo D, Lam SK (1999) J Catal 183:368–372
Maruska HP, Ghosh AK (1978) Sol Energy 20:443–458
Bickley R, Gonzalez-Carreno T, Lees J, Palmisano L, Tille R (1991) J Solid State Chem 92:178–190
Yu J, Yu J, Ho W, Jiang Z (2002) New J Chem 26:607–613
Okada K, Yamamoto N, Kameshima Y, Yasumori A, MacKenzie K (2001) J Am Ceram Soc 84:1591–1596
Ohtsuka Y, Fujiki Y, Suzuki Y (1982) J Jpn Assoc Mineral Petrol Econ Geol 77:17–124
Iida Y, Ozaki S (1961) J Am Ceram Soc 44:120–127
Chao HE, Yun YU, Xingfang HU, Larbot A (2003) J Eur Ceram Soc 23:1457–1464
Martinez J, Acosta DR, Cedillo G (2005) Thin Solid Films 490:118–123
Akurati KK, Vital A, Hany R, Bommer B, Graule T, Winterer M (2005) Int J Photoenergy 7:153–161
Yang J, Li D, Wang X, Yang L, Lu L (2002) J Solid State Chem 165:193–198
Mahanty S, Roy S, Sen S (2004) J Crystal Growth 261:77–81
Shi ZM, Yan L, Jin LN, Lu XM, Zhao G (2007) J Non-Cryst Sol 353:2171–2178
Baker RW (2004) Membrane technology and application. Wiley, Chichester
Guo B, Liu Z, Hong L, Jiang H (2005) Surf Coating Technol 198:24–29
Verma A, Samanta SB, Bakhshi AK, Agnihotry SA (2005) Sol Energy Mater Sol Cells 88:47–64
Fresno F, Coronado JM, Tudela D, Soria J (2005) Appl Catal B Environ 55:159–167
Tai WP, Oh JH (2002) Sens Actuators B 85:154–157
Kumar KN, Fray DJ, Nair J, Mizukami F, Okubo T (2007) Scripta Mater 57:771–774
Xu YL (1991) The basics of semiconducting oxides and compounds. Press of Xi’an Electronic Science and Technology University, Xi’an
Klug P, Alexander LE (1974) X-Ray diffraction procedures. Wiley, New York
Spurr RA, Myers H (1957) Anal Chem 29:760–762
Sayılkana F, Asiltürka M, Kirazb N (2009) J Hazard Mater 162:1309–1316
Hirata T, Ishioka K, Kitajima M, Doi H (1996) Phys Rev B 53:8442–8448
Sanjines R, Tang H, Berger H, Gozzo F, Margaritondo G (1994) J Appl Phys 75:2945–2951
Colmenares JC, Aramendia MA, Marinas A, Marinas JM, Urbano FJ (2006) Appl Catal A Gen 306:120–127
Shi LY, Gu HC, Li CZ, Fang D, Zhang Y, Hua B (1999) Chin J Catal 20:338–342
Lia XZ, Li FB, Yang CL, Ge WK (2001) J Photochem Photobiol A141:209–217
Li XZ, Li FB (2001) Environ Sci Technol 35:2381–2387
Garadkar KM, Patil AA, Hankare PP, Chate PA, Sathe DJ, Delekar SD (2009) J Alloys Comp 487:786–789
Zheng SK, Wang TM, Hao WC, Shen R (2002) Vacuum 65:155–159
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kaleji, B.K., Sarraf-Mamoory, R., Nakata, K. et al. The effect of Sn dopant on crystal structure and photocatalytic behavior of nanostructured titania thin films. J Sol-Gel Sci Technol 60, 99–107 (2011). https://doi.org/10.1007/s10971-011-2560-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-011-2560-2