
Abstract
Combination of the surfactant-free nonaqueous sol–gel approach with the microwave technique makes it possible to synthesize Fe3O4, CoFe2O4, MnFe2O4, and NiFe2O4 nanoparticles of about 5–6 nm and with high crystallinity and good morphological uniformity. The synthesis involves the reaction of metal acetates or acetylacetonates as precursors with benzyl alcohol at 170 °C under microwave irradiation of 12 min. Immersion of glass substrates in the reaction solution results in the deposition of homogeneous metal ferrite films whose thickness can be adjusted through the precursor concentration. If preformed nickel nanoparticles are used as a type of curved substrate, the ferrite nanoparticles coat the seeds and form core–shell structures. These results extend the microwave-assisted nonaqueous sol–gel approach beyond the simple synthesis of nanoparticles to the preparation of thin films on flat or curved substrates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The study of size dependent properties is a major driving force behind nanoparticle research, and size also plays a dominant role in determining the magnetic properties of nanoparticles [1–3]. Nanoparticles such as magnetite Fe3O4 and ferrites MFe2O4 (M = Mn, Co, Ni, etc.) became an important class of materials with unique magnetic, magnetoresistive and magneto-optical properties. They gave rise to a wide range of technological applications in magnetic recording and catalysis, but also in biomedical fields such as magnetic resonance imaging contrast agents or magnetic carriers for drug targeting [4–8]. One of the special features is the possibility to systematically tune their magnetic properties by varying the composition (by the type of the M2+ cation) or by changing the crystal morphology, i.e., size and shape. Magnetic spinels of the Fe3O4 and MFe2O4 type have been successfully produced by various methods ranging from mechanical to gas phase and solution routes [9–12]. The latter include co-precipitation, thermal decomposition, hydro- and solvothermal, and reverse micelle approaches [13–25]. Whereas co-precipitation is particularly suitable for biological applications due to the formation of nanoparticles in aqueous media, thermal decomposition or hydrothermal methods are good alternatives in terms of precise morphology control.
Without any doubts, nanoparticle synthesis has made immense progress in the last few years. Remaining challenges that have to be faced are the implementation of ecological and economic factors in the synthesis protocols. Modern synthesis routes have to be energy efficient, i.e., higher yields and higher purities at less energy consumption. Additionally, toxic solvents and additives should be avoided. In this regard, microwave chemistry seems to be particularly promising and has recently been applied to the synthesis of ferrite nanoparticles, too [26]. Surfactant-free nonaqueous sol–gel approaches were extremely successful in the preparation of nanocrystalline metal oxides [27, 28]. In combination with microwave heating these routes provide a fast and energy efficient synthesis methodology to metal oxide nanoparticles. As a matter of fact, a great variety of nanoparticles, including ZnO [29–31], doped zinc oxide [32, 33], CoO [30], MnO/Mn3O4 [30] or BaTiO3 [30] are accessible in a very controlled manner at moderate temperatures using microwave-assisted nonaqueous sol–gel chemistry. Microwave heating is beneficial for nonaqueous sol–gel chemistry in many ways. Most strikingly, typical reaction times for nanoparticle formation are reduced from days to minutes. In addition, microwave irradiation results in direct volumetric and, in the case of small reaction vessels, very homogeneous heating with reduced wall effects [34]. These conditions are ideal for the formation of monodispersed colloids, which require a fast and short nucleation burst, separated from the following growth stage [9]. Taking into account that the quality (i.e., high crystallinity, uniform crystal size and shape as well as small size distribution) of the final nanoparticles is highly dependent on the initial conditions and on the homogeneity of the reaction, it is essential to have an efficient heating tool.
The deposition of nanoparticles on specific substrates is another critical step in the development of devices exploiting their unique properties. Thin film technology making use of semiconducting, metallic, insulating or optical properties is omnipresent in our daily lives and plays a major role in almost all areas of industrial applications. There are numerous chemical and physical methods available to deposit materials in the form of thin films, including atomic layer deposition [35], molecular beam epitaxy [36] or magnetron sputtering [37]. However, thin film formation has also been addressed by sol–gel chemistry. In this case, film processing involves a dip- or spin-coating step, during which the inorganic sol is deposited onto a substrate [38]. Having this processing technique in mind, it is important to know that microwave irradiation provides a selective heating tool, which means that substances with different microwave absorbing properties will be unequally heated. For example, the surface of oxides covered by hydroxyl groups is more susceptible to absorb microwave energy than the bulk, leading to selective surface heating [39]. Accordingly, it should be possible to take advantage of microwave irradiation to selectively activate the surface of a substrate, whether it is a flat substrate or a curved particle, and to deposit another material on top of it. As a matter of fact, several papers reported the microwave-assisted solution deposition of metal oxides such as indium tin oxide [40], ZnO [41], TiO2 [42], BaWO4 [43], or Eu:YVO4 [44], on conducting glass substrates or silicon. Also core–shell nanoparticles have been prepared in the microwave by coating preformed nanoparticles with another material, e.g., CdTe/CdS [45], or Au@Ag [46].
In this paper we report the simultaneous formation of ferrite nanoparticle dispersions and deposition of thin films on glass substrates. The synthesis is based on a combination of nonaqueous sol–gel chemistry with the microwave technique, involving the reaction of metal acetates and acetylacetonates with benzyl alcohol [28]. This simple procedure reduces the “classical” sequential process for thin film preparation, i.e., synthesis of crystalline nanoparticles, preparation of stable dispersions, and deposition of films by dip- or spin-coating, into just one step. The composition of the ferrite nanoparticles can be varied from Fe3O4 to CoFe2O4, MnFe2O4 and NiFe2O4, and the small crystal sizes below 10 nm together with a narrow size distribution lead to superparamagnetic behavior. In addition to controlling the composition, the method offers the possibility of changing the crystal size by simply varying the experimental parameters like temperature and time. The use of benzyl alcohol, well known to be a low toxicity solvent [47], offers biocompatible conditions and coupled with microwave heating provides an economic, efficient and easily scaled up tool for the synthesis of magnetic nanoparticles. Beside the formation of nanoparticle dispersions, it is also possible to achieve homogeneous films with thicknesses of a few tens of nanometers within minutes by placing a glass substrate in the reaction solution. Moreover, the process can be extended to core–shell nanoparticles. Preliminary results on the coating of preformed nickel nanoparticles with the ferrites revealed that this technique is suitable for any size or shape of substrates giving rise to uniform coverage with thickness and composition dependent on the reaction parameters.
2 Experimental procedures
2.1 Materials
Fe(III) acetylacetonate ≥99.9%, Fe(II) acetylacetonate, Co(II) acetylacetonate 97%, Ni(II) acetate tetrahydrate 99.998%, and Mn(II) acetate 98% were used as precursors and anhydrous benzyl alcohol 99.8% as solvent. All chemicals were purchased from Sigma–Aldrich, stored under argon atmosphere and used as received. Glass slides 18 × 18 mm (Menzler-Gläser) were broken in half, washed in diluted HCl and sonicated.
2.2 Synthesis of Fe3O4 nanoparticles
About 1 mmol of iron(III) acetylacetonate was dissolved in 5 ml of benzyl alcohol in a glovebox under inert atmosphere (O2 and H2O < 0.1 ppm) and transferred into a 10 ml glass tube, sealed with a Teflon cap. The reaction mixture was heated at a temperature of 170 °C and an irradiation time of 12 min. After the reaction, the solution is thermally quenched by compressed air. The resulting suspensions were separated from the liquid phase by centrifugation and the precipitate washed with ethanol and diethyl ether and left to dry. The powders obtained were ground in a mortar.
2.3 Synthesis of MFe2O4 nanoparticles
The nanoscale transition metal ferrites were obtained by simultaneous reaction of Fe3+ and M2+ precursors in benzyl alcohol under similar experimental conditions as described for iron oxide. About 0.5 mmol of iron(III) acetylacetonate was transferred in a 10 ml glass tube followed by the addition of stoichiometric quantities of manganese(II) acetate, cobalt(II) acetylacetonate or nickel(II) acetate tetrahydrate.
2.4 Synthesis of Ni@Fe3O4
About 1 mmol of nickel(II) acetate was dissolved in 5 ml of benzyl alcohol, transferred in a sealed 10 ml glass tube and heated at 180 °C for 5 min. Once cooled, 1 mmol of iron(II) acetylacetonate was placed in the reaction medium and heated again at 180 °C for 5 min.
2.5 Microwave heating protocol
During a typical run the power is adjusted to heat the sample up to 170 °C, and then the temperature is kept for 12 min with high stirring rate to allow a good dispersion of the precursors in benzyl alcohol. The reaction is performed in a sealed vessel of 10 ml with magnetic stirring and under microwave irradiation. The temperature and the pressure are controlled by an IR thermometer and a pressure sensor, respectively.
2.6 Dispersions
Dispersions containing 0.2% w/v of the as-synthesised nanopowders in ethanol were prepared as follows. The organic liquid was removed from the reaction mixture by centrifugation. The precipitate was twice washed with ethanol and then, without drying, dispersed again using sonication in the corresponding amount of ethanol.
2.7 Film preparation
Standard microscopy glass slides were soaked into hydrochloric acid solutions overnight, washed in deionised water and dried in air. Films were prepared by immersing the glass slides into the reaction solution containing Fe3+ (and Co2+) precursors in total concentrations of 0.005–0.15 M in benzyl alcohol. Stirring is omitted to avoid damaging of the glass slide. The samples are heated up to 170 °C, and then the temperature is kept for 12 min.
2.8 Instruments and characterization
The microwave experiments were carried out by using a CEM Discover reactor operating at a frequency of 2.45 GHz. X-ray powder diffraction studies (XRD) were performed on a Philips PW 1800 diffractometer in reflection mode using Cu Kα λ = 1.5406 Å radiation and a post-sample monochromator. The particle size, d, was determined from the peak broadening by Scherrer’s equation \( d = k\lambda /(\beta \cos \theta \)) (where β is the peak FWHM measured in radians and θ is the Bragg angle and k = 0.9). Transmission electron microscopy (TEM) and high resolution transmission electron microscopy were performed either on a Philips CM30ST (LaB6 cathode, operated at 300 kV, point resolution of 2 Å) or on a Philips CM200-FEG (200 kV, Cs = 1.35 mm) microscope. The latter was used for energy-dispersive X-ray spectroscopy (EDX). The samples were ground and then suspended in ethanol. One drop of this suspension was deposited on a 400-mesh carbon-coated copper grid. To minimise agglomeration of the nanoparticles the copper grid was placed on a filter paper. AFM measurements were carried out in contact mode with a WITec Mercury 100 AFM using a silicon nitride tip. The vertical resolution is about 2 nm, whereas the lateral resolution is 5 nm. XPS was performed on a Sigma Probe (Thermo Scientific), using an Al Kα source operated at 200 W. Photoelectrons were detected with a hemispherical analyzer at a pass energy of 25 eV for the detail and 100 eV for the survey spectra and a take off angle of 90°. Data were analyzed using the CasaXPS software (CasaXPS software Version 2.3.15dev52, Software Ltd). A Shirley background was subtracted before the peak areas were integrated and corrected for the cross section using the Scofield factors [48], inelastic mean free path, attenuation length [49] and the energy dependent transmission function. To account for charging effects, the binding energy of the C1 s peak was normalized to 285.0 eV. The Fe2p3/2 peak was fitted with three peaks assigned to FeOOH (BE = 711.6 eV, FWHM = 3.4 eV), Fe3+ (BE = 710.5 eV, FWHM = 2.4 eV) and Fe2+ (BE = 709.0 eV, FWHM = 2.4 eV) with a Gaussian:Lorenzian ratio of 45:55 [50]. UV/Vis spectra were recorded at room temperature with a Perkin Elmer Lambda 20. The microstructure of the thin films was characterized using scanning electron microscopy (SEM, Leo 1530, Germany). The top layers of the thin films were sputtered (Bal-Tec, SCD 050, Sputter Coater) with a platinum coating to allow for imaging at higher resolutions. Magnetic properties were measured using a superquantum interference design (SQUID) magnetometer MPMS 5S in the temperature range of 2–300 K and a field of 0–5 T. Magnetic susceptibilities were measured between 2 and 300 K in an external magnetic field of H = 1000 Oe. Magnetization curves were obtained at T = 300 K.
3 Results and discussion
3.1 Ferrite nanoparticle synthesis and dispersions
As illustrated in Fig. 1a, the microwave-assisted nonaqueous sol–gel route can conveniently be extended to various iron oxide spinels by reacting Fe(III) acetylacetonate and transition metal acetates or acetylacetonates with benzyl alcohol. Already at temperatures as low as 170 °C uniform and nearly spherical nanocrystallites with the composition MFe2O4 (M = Fe2+, Co2+, Mn2+, Ni2+) are accessible. The as-synthesized nanoparticles can easily be isolated from the organic side products and from residual precursors by centrifugation and washing with ethanol and diethyl ether. After drying and grinding, dark brownish to black powders are obtained, whose magnetic properties are visible by naked eye upon responding to a permanent magnet (Fig. 1b). In addition to the powder, stable transparent dispersions of nanoparticles in ethanol are formed after sonicating the washing liquid during the second washing step (Fig. 1c). Similarly, the process can be used for thin film deposition, as shown in Fig. 1d. The mildly etched glass slides were placed directly in the precursor solution. After microwave heating for 12 min, the glass slides turned brownish, indicating the deposition of ferrite films on the surface. The films were rinsed with ethanol and dried under ambient conditions. This technique is not restricted to flat substrates, but can be extended to core–shell structures, in which preformed metal nanoparticles play the role of a curved substrate. Fe3O4 nanoparticles, supported on the surface of Ni, were obtained by having preformed nickel nanoparticles in the initial Fe3O4 precursor solution. The metallic Ni nanoparticles were obtained by irradiating Ni(II) acetate in benzyl alcohol at 180° C for 5 min [51].

a General reaction scheme displaying the metal oxide precursors used, the solvent, the experimental conditions, and the final composition of the nanoparticles. Photographs of b magnetite nanopowder under the influence of a permanent magnet, c CoFe2O4 dispersion in ethanol, and d CoFe2O4 film on a glass substrate
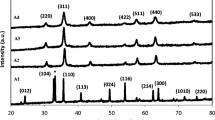
The phase and crystallinity of the different powders were investigated by powder X-ray diffraction (XRD), as shown in Fig. 2. All the patterns (A Fe3O4, B CoFe2O4, C MnFe2O4 and D NiFe2O4) correspond to just one crystalline phase with the inverse spinel structure. Accordingly, they can be indexed to magnetite (ICDD PDF No. 1-1111), cobalt ferrite (ICDD PDF No. 1-1121), manganese ferrite (ICDD PDF No. 10-0319) and nickel ferrite (ICDD PDF No. 3-0875), respectively. The broad reflections observed are characteristic of small nanocrystals. The crystallite sizes calculated from the 400 and 440 reflections using the Scherrer equation were estimated to be in the range 5–6 nm for all ferrites.

XRD pattern of a Fe3O4, b CoFe2O4, c MnFe2O4 and d NiFe2O4 nanoparticles. Vertical doted lines correspond to the peak position of cubic Fe3O4 [ICDD PDF 1-1111]
The size and shape of the synthesized MFe2O4 nanocrystals were investigated using transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM). A drop of the transparent brownish ethanol solution, as described in the experimental section, is deposited on a carbon coated copper grid. Figure 3 shows representative overview images of A) CoFe2O4 C) MnFe2O4 and D) NiFe2O4. All the ferrites exhibit a nearly spherical (i.e., isotropic) shape, a low degree of agglomeration and a narrow size distribution typically in the range 4–8 nm. These results underline the importance of microwave irradiation in reducing the agglomeration of the nanoparticles by a fast heating, thus allowing the preparation of stable dispersions without the use of any surfactants. The TEM images confirm that the particles are well below 10 nm in size, which is in good agreement with the XRD data. Figure 3b shows a HRTEM image of CoFe2O4. The clearly visible lattice fringes give additional evidence for the high crystallinity of the nanoparticles. The chemical composition was investigated by energy-dispersive X-ray spectroscopy (EDX) and is summarized in Table 1. The different MFe2O4 nanoparticles display a ratio of M/Fe of about 0.5. To evaluate the compositional homogeneity, we carried out several EDX analyses at different locations of the samples. Deviations of less than 0.05% support the high compositional uniformity. The oxidation states of the different transition metals in the MFe2O4 nanocrystals were elucidated by XPS (Fig. 4). The 2p3/2 binding energy of Co (780.1 ± 0.3 eV) and its satellite around 785.7 eV, characteristic for Co2+ [52–54], the binding energy of Mn (641.1 ± 0.1 eV) and Ni (854.4 ± 0.2 eV) [53, 55, 56], and the Ni 2p1/2 peak shape [52] indicate that all these transition metals are twofold positively charged, as expected for a spinel structure. Furthermore, iron is predominantly in a Fe3+ oxidation state as indicated by the Fe 2p3/2 peak which is centered at 710.5 eV and the satellite peak around 719 eV characteristic for Fe3+ [57]. Deconvolution of the Fe 2p3/2 peak of magnetite, centered at 710.2 eV into three peaks assigned to Fe3+ (710.5 eV), Fe2+ (709.0 eV) and FeOOH (711.6 eV), respectively, reveals a molar ratio of Fe3+ to Fe2+ of 0.6. Furthermore, with time, magnetite oxidized to Fe2O3, which was reflected in a shift of the Fe 2p3/2 peak towards 710.5 eV and the appearance of a more pronounced Fe3+ satellite peak. The Fe3+ satellite peak is convoluted with the Fe2+ satellite peak for freshly synthesized magnetite nanoparticles (Fig. 4d).

TEM overview images of a CoFe2O4, c MnFe2O4 and d NiFe2O4 nanoparticles. b HRTEM image of CoFe2O4

XPS spectra of the as-synthesized nanopowders. a Co 2p of CoFe2O4, b Mn 2p of MnFe2O4, c Ni 2p of NiFe2O4. d Fe 2p of a) CoFe2O4, b) MnFe2O4, c) NiFe2O4 and d) Fe3O4 nanoparticles
The magnetic properties of the different MFe2O4 powders were measured by a Quantum Design SQUID magnetometer. Figure 5 shows the magnetization curves at 300 K. At H = 50 kOe the magnetization is found to be 19.7, 30.6, 47.0 and 48.0 emu/g for MnFe2O4 (Fig. 5a), CoFe2O4 (Fig. 5b), Fe3O4 (Fig. 5c) and NiFe2O4 (Fig. 5d), respectively. The observed values are within the range reported in the literature for such nanoparticles [16, 18, 25, 58]. The temperature dependence of the magnetization was measured using zero field cooling and field cooling (ZFC–FC) procedures from 2 to 300 K in an applied field of 1000 Oe (Fig. 6). With increasing temperature the ZFC magnetization increases, before reaching a maximum value, i.e., the blocking temperature T B , at about 40 K for Fe3O4 (Fig. 6a), 146 K for CoFe2O4 (Fig. 6b), 25 K for MnFe2O4 (Fig. 6c) and 19 K for NiFe2O4 (Fig. 6d). At temperatures higher than T B , the thermal energy is larger than the magnetic energy barrier and the material becomes superparamagnetic. The FC magnetization increases steadily from 300 to 2 K and deviates from the ZFC curve below T B . Field-dependent magnetization measurements show low coercivities with no remanence giving additional proof for the superparamagnetic behaviour.

Magnetization curves measured at 300 K for a MnFe2O4, b CoFe2O4, c Fe3O4 and d NiFe2O4 powder

Temperature dependence of the magnetization at 1000 Oe for a Fe3O4, b CoFe2O4, c MnFe2O4 and d NiFe2O4 powders
3.2 Thin film deposition
Morphological studies of the as-deposited CoFe2O4 thin films were performed by SEM and AFM. The CoFe2O4 layers were prepared from reaction mixtures with various total precursor concentrations ranging from 0.005 to 0.15 M. Figure 7a (overview image at low resolution) and 7b (higher magnification) display the SEM results obtained for 0.005 M. The surface topology is quite smooth and homogeneous over the whole area, although a few spherical agglomerates are visible, too (Fig. 7a). On the other hand, the layer synthesized at 0.15 M (Fig. 7c, d) exhibits a more inhomogeneous morphology with the presence of microcracks. The AFM images (Fig. 7e, f) complement the SEM study, allowing an estimation of the layer thickness to be around 80 nm for an initial condition of 0.15 M total precursor concentration (Fig. 7f). Upon decreasing the total precursor concentration to 0.005 M, a step height of about 25 nm could be measured (Fig. 7e). Section analysis gives evidence of the film homogeneity with a low surface roughness.

a–d SEM images of CoFe2O4 films prepared at 0.005 M total precursor concentration (a and b) and at 0.15 M (c and d). Corresponding AFM measurements e 0.005 M and f 0.15 M
XPS spectra of as-deposited CoFe2O4 films (Fig. 8) were well in agreement with those measured on CoFe2O4 powder. The survey spectrum of these films (Fig. 8a) revealed carbon as the only impurity present in the films. Si from the underlying substrate could not be detected. Obviously, the deposited CoFe2O4 film is thicker than 10 nm, which is the XPS sampling depth. It can be seen that neither cobalt nor iron underwent changes in the oxidation state during film deposition (Fig. 8b, c).

XPS spectra of CoFe2O4 films deposited on SiO2 glass. a Survey spectrum, b Co 2p and c Fe 2p spectra
The optical spectrum of cobalt ferrite nanoparticles is generally dominated by an absorption in the range of 300–350 nm [59]. A series of experiments were performed to monitor the film thickness evolution by UV/Vis spectroscopy. The absorbance of the films deposited on glass substrates were measured in the wavelength range 300–1000 nm by subtracting the absorbance of the glass substrate, which was taken as a reference. As shown in Fig. 9, the absorbance in the 300–350 nm range increased with increasing concentration (0.005–0.15 M) as expected. The increase of the absorbance at 320 nm with the reaction concentration can be linearly fitted, indicating that the reaction conditions, i.e., the precursor concentration, directly affect the layer thickness (Fig. 9, inset). Measurements performed at different positions of the layers gave similar absorbance values, suggesting that the deposition is quite uniform. These are perfect conditions for a precise control of the layer thickness simply by adjusting the experimental conditions.

UV/Vis spectra of CoFe2O4 films obtained with different initial precursor concentrations ranging from 0.005 (bottom curve) to 0.15 M (top curve). Inset: Linearly fitted trend of A(λ=320nm) versus solution concentration. In all cases the spectra have been corrected for the absorption of the bare glass substrate
3.3 Ni@Fe3O4 synthesis
Ni@Fe3O4 core–shell nanoparticles were prepared via the deposition of Fe3O4 on preformed Ni particles. Figure 10a shows a TEM overview image of such a Ni@Fe3O4 configuration with larger Ni nanoparticles as core (dark contrast) and smaller Fe3O4 nanoparticles as surrounding shell. The XRD pattern in Fig. 10b indicates that the as-prepared nanoparticles are highly crystalline, consisting of face-centred cubic nickel (JCPDS file No 04-0850) and cubic magnetite (ICDD PDF No. 1-1111). The particle sizes determined from the Scherrer equation are 20 nm from the 200 reflexion of Ni and 7 nm from the 220 reflexion of Fe3O4 and agree well with the TEM results.

a TEM image of metallic Ni in Fe3O4 dispersions and b the corresponding XRD pattern with cubic Ni [ICDD PDF 4-0850] and * Fe3O4 [ICDD PDF 1-1111]
4 Conclusions
The results presented here demonstrate the great potential of combining the microwave technique with nonaqueous sol–gel chemistry not only to produce various transition metal ferrite nanoparticles with high crystallinity and morphological uniformity, but also homogeneous metal ferrite films on flat and curved substrates. The film thickness on glass substrates can easily be varied in the range of about 20–80 nm by adjusting the precursor concentration. The fact that the ferrite nanoparticles can also be deposited on preformed metal nanoparticle seeds opens up new prospects for the preparation of multi-component core–shell materials.
References
Lu AH, Salabas EL, Schuth F (2007) Angew Chem Int Ed 46:1222–1244
Jeong U, Teng XW, Wang Y, Yang H, Xia YN (2007) Adv Mater 19:33–60
Darling SB, Bader SD (2005) J Mater Chem 15:4189–4195
Sugimoto M (1999) J Am Ceram Soc 82:269–280
Mornet S, Vasseur S, Grasset F, Veverka P, Goglio G, Demourgues A, Portier J, Pollert E, Duguet E (2006) Prog Solid State Chem 34:237–247
Reimer P, Weissleder R, Lee AS, Wittenberg J, Brady TJ (1990) Radiology 177:729–734
Alexiou C, Arnold W, Klein RJ, Parak FG, Hulin P, Bergemann C, Erhardt W, Wagenpfeil S, Lubbe AS (2000) Cancer Res 60:6641–6648
Jun YW, Seo JW, Cheon A (2008) Acc Chem Res 41:179–189
Park J, Joo J, Kwon SG, Jang Y, Hyeon T (2007) Angew Chem Int Ed 46:4630–4660
Cushing BL, Kolesnichenko VL, O’Connor CJ (2004) Chem Rev 104:3893–3946
Strobel R, Pratsinis SE (2007) J Mater Chem 17:4743–4756
Sepelak V, Bergmann I, Kipp S, Becker KD (2005) Z Anorg Allg Chem 631:993–1003
Kang E, Park J, Hwang Y, Kang M, Park JG, Hyeon T (2004) J Phys Chem B 108:13932–13935
Hyeon T, Lee SS, Park J, Chung Y, Bin Na H (2001) J Am Chem Soc 123:12798–12801
Shemer G, Tirosh E, Livneh T, Markovich G (2007) J Phys Chem C 111:14334–14338
Jia X, Chen DR, Jiao XL, He T, Wang HY, Jiang W (2008) J Phys Chem C 112:911–917
Sun SH, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li GX (2004) J Am Chem Soc 126:273–279
Bao NZ, Shen LM, Wang YH, Padhan P, Gupta A (2007) J Am Chem Soc 129:12374–12375
Song O, Zhang ZJ (2004) J Am Chem Soc 126:6164–6168
Jana NR, Chen YF, Peng XG (2004) Chem Mater 16:3931–3935
Calero-DdelC VL, Rinaldi C (2007) J Magn Magn Mater 314:60–67
Zhao LJ, Zhang HJ, Xing Y, Song SY, Yu SY, Shi WD, Guo XM, Yang JH, Lei YQ, Cao F (2008) J Solid State Chem 181:245–252
Wu JH, Ko SP, Liu HL, Kim S, Ju JS, Keun-Kim Y (2007) Mater Lett 61:3124–3129
Pinna N, Grancharov S, Beato P, Bonville P, Antonietti M, Niederberger M (2005) Chem Mater 17:3044–3049
Yanez-Vilar S, Sanchez-Andujar M, Gomez-Aguirre C, Mira J, Senaris-Rodriguez MA, Castro-Garcia S (2009) J Solid State Chem 182:2685–2690
Wang WW (2008) Mater Chem Phys 108:227–231
Djerdj I, Arcon D, Jaglicic Z, Niederberger M (2008) J Solid State Chem 181:1571–1581
Pinna N, Niederberger M (2008) Angew Chem Int Ed 47:5292–5304
Hu XL, Gong JM, Zhang LZ, Yu JC (2008) Adv Mater 20:4845–4850
Bilecka I, Djerdj I, Niederberger M (2008) Chem Commun 886–888
Bilecka I, Elser P, Niederberger M (2009) ACS Nano 3:467–477
Hammarberg E, Prodi-Schwab A, Feldmann C (2009) J Colloid Interface Sci 334:29–36
Fidelus J, Piticescu RR, Piticescu RM, Lojkowski W, Giurgiu L (2008) Z Naturforsch B Chem Sci 63:725–729
Kappe CO, Dallinger D (2009) Mol Divers 13:71–193
Clavel M, Rauwel E, Willinger M-G, Pinna N (2009) J Mater Chem 19:454–462
Chambers SA (2008) J Phys Condens Matter 20:264004
Han JG (2009) J Phys D Appl Phys 42:043001
Brinker CJ, Hurd AJ, Schunk PR, Frye GC, Ashley CS (1992) J Non-Cryst Solids 147:424–436
Vallee SJ, Conner WC (2008) J Phys Chem B 112:15483–15489
Okuya M, Ito N, Shiozaki K (2007) Thin Solid Films 515:8656–8659
Peiro AM, Domingo C, Peral J, Domenech X, Vigil E, Hernandez-Fenollosa MA, Mollar M, Mari B, Ayllon JA (2005) Thin Solid Films 483:79–83
Vigil E, Saadoun L, Ayllon JA, Domenech X, Zumeta I, Rodriguez-Clemente R (2000) Thin Solid Films 365:12–18
Wang R, Liu C, Zeng J, Li KW, Wang H (2009) J Solid State Chem 182:677–684
Xu HY, Wang H, Jin TN, Yan H (2005) Nanotechnology 16:65–69
He Y, Lu HT, Sai LM, Lai WY, Fan QL, Wang LH, Huang W (2006) J Phys Chem B 110:13370–13374
Tsuji M, Miyamae N, Lim S, Kimura K, Zhang X, Hikino S, Nishio M (2006) Cryst Growth Des 6:1801–1807
Nair B (2001) Int J Toxicol 20:23–50
Scofield JH (1976) J Electron Spectros Relat Phenomena 8:129–137
Seah MP, Dench WA (1979) Surf Interface Anal 1:2–11
Olla M, Navarra G, Elsener B, Rossi A (2006) Surf Interface Anal 38:964–974
Jia FL, Zhang LZ, Shang XY, Yang Y (2008) Adv Mater 20:1050–1054
Kim JG, Pugmire DL, Battaglia D, Langell MA (2000) Appl Surf Sci 165:70–84
Allen GC, Harris SJ, Jutson JA, Dyke JM (1989) Appl Surf Sci 37:111–134
Chu YQ, Fu ZW, Qin QZ (2004) Electrochim Acta 49:4915–4921
McIntyre NS, Cook MG (1975) Anal Chem 47:2208–2213
Lenglet M, Hochu F, Durr J, Tuilier MH (1997) Solid State Commun 104:793–798
Brundle CR, Chuang TJ, Wandelt K (1977) Surf Sci 68:459–468
Zi ZF, Sun YP, Zhu XB, Yang ZR, Dai JM, Song WH (2009) J Magn Magn Mater 321:1251–1255
Bala T, Sankar CR, Baidakova M, Osipov V, Enoki T, Joy PA, Prasad BLV, Sastry M (2005) Langmuir 21:10638–10643
Acknowledgments
Financial support by ETH Zürich and the Swiss National Science Foundation (Project No. 200021_124632) is gratefully acknowledged. We thank Li Luo for SEM, Dr. Igor Djerdj for TEM, Barbara Grant for AFM, and Christian Mensing for the SQUID measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bilecka, I., Kubli, M., Amstad, E. et al. Simultaneous formation of ferrite nanocrystals and deposition of thin films via a microwave-assisted nonaqueous sol–gel process. J Sol-Gel Sci Technol 57, 313–322 (2011). https://doi.org/10.1007/s10971-010-2165-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-010-2165-1