
Abstract
Vanadium oxide/hexadecylamine (V2O5/HDA) sensing membrane was deposited on the glassy carbon substrate and used as the sensing layer of the extended gate H+-ion sensitive field effect transistor (EGFET) device. The structural and morphological features of V2O5/HDA were studied by X-ray diffraction, Fourier transformed-infrared spectroscopy and Scanning electronic microscopy images; and the electrochemical behavior was analyzed by cyclic voltammogram. V2O5/HDA presents a lamellar structure as well as several rod formations. The material stabilizes electrochemically after several cycles and leads to reproducibility of Li+ ion insertion/de-insertion into the vanadium oxide structure. The material was investigated as a pH sensor in the pH range 2–12 and presented a sensitivity of 38.1 mV/pH. The sensitive membrane structure is simple to fabricate and the measurement is fast for application as a disposable sensor.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Since Bergveld [1] employed the first field effect transistor, many papers have presented how to design and develop pH sensors [2–4]. Ion sensitive field-effect transistors (ISFETs) have been developed on the basis of MOSFET (metal oxide semiconductor field effect transistor) to measure pH and a variety of other ions [5]. The difference between ISFET and MOSFET is that there is no metal gate electrode, given the gate is directly exposed to the buffer solution [6]. As described by Chang K.-M. et al. [7], the measurement is made when there is a change in the surface potential between the gate insulator and the electrolyte, the electric field at the insulator semiconductor interface will be changed and the channel conductance that affects the drain current will also be modulated. Since the channel conductance and drain current can be modulated, it is possible to measure the changes by applying a fixed source to drain voltage. By means of this method, one can plot a standard linear line between gate voltages and various pH values, and a standard linear line can be taken to measure an unknown acid or an alkaline solution.
The development of ISFET has been going on over than 35 years, and the first sensitive membrane to be used was silicon dioxide (SiO2), which showed an unstable sensitivity and large drift. Several dielectric membranes, including Si3N4, Al2O3, SnO2 and Ta2O5 [1], have been used as pH-sensitive membranes because of their higher pH response as ISFET-pH sensor. Some problems of the ISFET configuration can be overcome by using the structure of the extended gate field effect transistor (EGFET). The flexible shape of the extended gate structure is another advantage of the EGFET. The EGFET has better long-term stability because ions from the chemical environment are excluded from any region close to the FET gate insulator [8]. The sensitive layered structure of the EGFET is fabricated on the end of the signal line extended from the FET gate electrode [9]. There are several kinds of ion-sensing membranes applied in the pH-sensing dielectric layers of pH-EGFET, such as ruthenium oxide [10], carbon nanotube [11], SnO2 [12], ZnO [13] and V2O5 xerogel [14]. In the search for other alternatives for ion-sensing membranes to be employed in pH sensors, one can point out to the vanadium oxide after thermal treatment. The importance of the development of this transition-metal is the fact that they present a large number of oxidation states with possible redox-active properties [15]. Actually, the electrochromism and electrochemical properties as well as applications in electrochemical sensors have been developed [16]. Another interesting subject related to vanadium oxide is the structural versatility, which allows insertion of a wide variety of guest species, such as polymers and surfactants [17, 18], and this combination is known as hybrid material [19, 20]. The combination of different organic/inorganic components in a single material may lead to unique electrical, optical, and mechanical properties that may not be present in the starting components alone. [21]. The synergic effect resulting from the combined properties of hybrid material can give rise to very interesting features, especially in what concerns electrochemical, electrochromic and conduction properties [22].
In this context, our interest is to investigate the electrical response of V2O5/HDA to fabricate a pH-EGFET sensor and to try to explore the interaction of the ion-sensitive membrane with charges in solution, find out the sensor sensitivity, and verify whether it is suitable as a pH sensor. Further on the electrical response as pH sensor, V2O5/HDA was characterized by various techniques, such as X-ray diffraction (XRD), infrared spectroscopy (FTIR), scanning electronic microscopy (SEM) and cyclic voltammetry (CV).
2 Experimental procedure
2.1 Synthesis
In a typical synthetic procedure, the molar ratio of V2O5/hexadecylamine was 1:1, which corresponded to 0.017 M (3.09 g) of crystalline V2O5 (Acros), which was slowly added to a beaker containing a transparent ethanolic solution (5.7 mL) of 0.017 M hexadecylamine (HDA) 90% (4.55 g, Aldrich). The mixture was maintained under vigorous stirring for 2 h. Then, 16.7 mL of purified water (Milli-Q–Millipore system) was added. This material was aged for 48 h. Finally, a hydrothermal reaction was carried out in a homemade autoclave apparatus at 180 °C for 7 days. The final product was washed with water and absolute ethanol and dried under vacuum.
2.2 Characterization
The X-ray diffraction (XRD) data were recorded on a SIEMENS D5005 diffractometer using a graphite monochromator and CuKα emission lines (1.541 Å, 40 kV, 40 mA). To this end, the sample (in the powder form) was deposited onto a glass plate and data were collected at room temperature over the range 2° ≤ 2θ ≤ 50°, with a resolution of 0.020°.
Fourier-transform infrared spectra (FTIR) were recorded from 4,000 to 400 cm−1 on a Bomem MB 100 spectrometer. The samples were dispersed in KBr and pressed into pellets.
Scanning electron microscopy (SEM) was carried out on a ZEISS microscope EVO 50 model operating at 20 kV. A thin gold coating (≈20 Å) was applied to the sample using a Sputter Coater—Balzers SCD 050. The material obtained after thermal treatment were measured by the Image Tool software.
Cyclic voltammograms were measured using an AUTOLAB (EcoChemie) model PGSTAT30 (GPES/FRA) potentiostat/galvanostat interfaced with a computer. The electrode arrangement consisted of a glassy carbon as the working electrode, a platinum wire auxiliary electrode, and saturated calomel electrode (SCE) as reference [22]. The V2O5/HDA was deposited, ~5 μL, on the electrode surface and dried at room temperature (25 °C). The supporting electrolyte 1.0 M LiClO4 was studied in acetonitrile medium. In addition, all the experiments were carried out in deoxigenated solutions by bubbling N2, at room temperature.
The electrical response of the sensor was measured using solutions of various pH values, and the curves were obtained by an Agilent 34970A parameter analyzer. To this end, ~5 μL of the V2O5/HDA was deposited on the glassy carbon substrate at room temperature (24 °C), to establish electric contact with the gate.
3 Results and discussion
FTIR measurements were carried out in order to characterize the material and verify whether the surfactant used during the synthesis was present. Figure 1 shows the FTIR spectrum of vanadium oxide and it is possible to observe the four characteristic bands of the vanadium oxide at 994, 748 and 550–450 cm−1, corresponding to the vibrational modes of the V5+ species in the vanadyl group (V=O), V–O–V asymmetric stretch, and V–O–V symmetric stretch mixed with the deforming vibrations of the vanadium oxygen polyhedra, respectively [23]. The band at 909 cm−1 in the spectrum of the vanadium oxide is close to that of α-V4+ which is considerably less ordered due to a distorted octahedron-shaped vanadyl unit-cell arrangement [24]. In order to remove the excess hexadecylamine, vanadium oxide was washed with water and absolute ethanol and dried under vacuum. It is possible to observe that the presence of the bands around 2,920, 2,840 and 1,467 cm−1 could be assigned to ν as(C–H), ν s(C–H), δ(CH2), vibration modes of CH3(CH2)15 NH2. Several bands, in weak transmittance, are observed between 3,400 and 3,500 cm−1, which can be related to symmetric out of plane angular deformation of N–H species from the surfactant, indicating that HDA can be present in the V2O5.

FTIR spectra of a V2O5 powder, b HDA and c V2O5/HDA
The X-ray diffraction pattern of V2O5 powder and V2O5/HDA are showed in Fig. 2(a) and (b), respectively. After the insertion of HDA, it was observed that the reaction leads to a crystalline product, which is a typical layered compound. It exhibits two series of reflections. The first is a 00ℓ set of reflections with high intensity (2θ < 10°), corresponding to the stacking of the layers perpendicular to the substrate, which is typical of layered structures (the 001 peak directly relates to the distance between the vanadium oxide interlayer). Therefore, the presence of the typical diffraction peaks (00ℓ) in the XRD patterns of vanadium oxide can also indicate the presence of the intercalated amine groups between these layers as observed in FTIR spectrum. The analysis of the 001 reflection indicates an increase in the interlayer spacing to 3.24 nm, which is consistent with the presence of the surfactant species in the vanadium oxide (d-spacing of 1.17 nm) [21]. The second set of reflections (h00) with lower intensity (2θ > 10°), corresponds to the two-dimensional structure of the layers, as also observed in the literature [25]. The 00ℓ series provides the distance between the vanadium oxide layers in lamellar distance with d 001, d 002, d 003 and d 004 values of 3.24, 1.60, 1.10 and 0.80 nm, respectively. It also shows that the HDA did not cause a collapse after insertion and the lamellar structure of the vanadium oxide was maintained. The characteristic h00 reflections, shown in Fig. 2, indicate high structural order of the vanadate layers. The plane lattice must come from a double sheet of vanadium atoms, containing vanadium mostly in distorted octahedral coordination [24, 26].

X-ray diffraction patterns of a V2O5 powder, b V2O5/HDA
The V2O5/HDA synthesis was carried out during 10 days, as described above. From the SEM image, it is possible to observe the arrangement of the rods. Figure 3a shows a very irregular surface after 3 days of synthesis indicating formation of few rods with diameter between 200 and 300 nm. Figure 3b displays the image of the oxide after 10 days of synthesis, showing that a rod arrangement took place with a diameter around 270 nm. The synthesis of this material can be occuring in two steps: in the first one, the intercalation of HDA ions in the lamellar precursor provokes a diminution of the van der Waals strengths between the layers; the second step is the hydrothermic treatment, where enough energy is supplied to the system provoking the rolling up process of layers (in order to maintain a minimum contact) [27]. In addition, Fig. 3c gives evidence that the rods are arranged almost parallel to each other.

SEM image of V2O5/HDA a after 3 days, b 10 days and c 10 days in another region
The electrochemical performance of V2O5/HDA as cathodic membrane was carried out by cyclic voltammetry (CV). The cyclic voltammograms of the V2O5/HDA material displays peaks in the potential range −0.5 to +1.5 V (SCE), in 0.1 mol/dm3 LiClO4 in acetonitrile solution, which were obtained at a scan rate of 20 mV s−1. There is one cathodic peak at +0.79 V and one anodic peak in −0.18 V (Fig. 4). These peaks correspond to the VV/IV redox pair with concomitant occurrence of lithium ion insertion/de-insertion as a consequence of vanadium oxidation/reduction process (xe− + xLi+ + V2O5 ⇆ LixV2O5). In addition, the width and asymmetric pattern of the peak can be related to the heterogeneity of the film surface, suggesting a more difficult lithium diffusion to reach active sites more internal into the film [21]. In a series of voltammetric scans between −0.5 and 1.5 V (Fig. 5), the cathodic peak was broadened and slightly shifted toward more negative (cathodic scans) potentials over 50 cycles. This result could be explained by modifications in the structure of the vanadium oxide sheets, which allow intercalation/deintercalation of more lithium ions in different sites, thus compensating for the charge in the film [26, 28]. However, the presence of HDA into the V2O5 causes an expansion between the layers and, as result, can facilitates the electromigration and accommodation of lithium ions during the intercalation and deintercalation processes.

Cyclic voltammograms of V2O5/HDA in 0.1 M LiClO4 in acetonitrile medium and ν = 20 mV/s

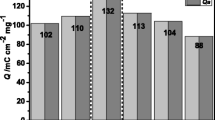
Total charge as a function of the number cycles of V2O5/HDA
The cyclic voltammograms of the V2O5/HDA shows that the first cycle presents a total charge value of 107 mC cm−2, which decreases to 59 mC cm−2 in the 25th cycle (a decrease of 55% in relation to the first cycle). However, it is possible to note that the decrease in total charge from the 25th cycle to the 50th (48 mC cm−2) is 18%. Theses values indicate that the vanadium oxide material exhibits reasonable electrochemical stability, since the percent total charge values very little after the 25th cycle. This can be associated with the presence of the rods and the presence of surfactant, which open the possibility of improving the stability of the vanadium framework. In addition, the presence of these rods enables improvement of the Li+ insertion/deinsertion process, causing an increase in the voltammetric stability as a function of the total charge leading to structural accommodation after several cycles, resulting in better performance. This characteristic can also influence the electrical behavior and, consequently, promote a good sensitivity to the vanadium oxide as sensing membrane.
Before the electrical behavior of the V2O5/HDA membrane as pH sensor was investigated, a study of the current variation as a function of time was performed. Figure 6 shows that there is a large variation in current until 150 s. After 250 s, the current does not change significantly. Additionally, the current interval between pH 2 and 12 is 27 μA at 0 s. After 300 s this interval increases to 46 μA indicating that time can influence the final results. Therefore, the sensitivity studies were measured after the V2O5/HDA had been submerged in the buffer solutions for 300 s. The different shapes of the curves in Fig. 6 are due to the fact that each pH solution is composed of different constituent ions (buffer solutions).

Current variation as a function of time for V2O5/HDA
Figure 7 shows drain-current, I D, as a function of the gate-source voltage, V GS, while source-drain voltage, V DS, is kept at 0.3 V. There is a clear shift toward higher voltages as a function of increasing of pH values. The sensitivity value (Fig. 8) was obtained from the relationship between V GS as a function of pH (for I DS fixed at 200 μA) from Fig. 7. As a result, the sensitivity of the sensor calculated, from a linear fit was 38.1 mV/pH, and it presented a good linearity. From the Nernstian law, it is known that the theoretical value, [29] is 59.2 mV/pH, so our experimental arrangement presented a reasonable response (deviation of 35%).

Response of a V2O5/HDA pH-EGFET sensor in the linear region when immersed into solutions with pH values ranging from 2 to 12

Sensitivity of the pH sensor: saturation response from pH 2 up to pH 12 in the linear region
In general, the sensitive membrane structure is easily fabricated and the measurement is simple for application as a disposable sensor. However, the experimental conditions, such as polymer insertion, must be improved so that values closer to the theoretical one are achieved. The device might be further tested as a urea and glucose sensor.
4 Conclusion
The synthesis, structural and electrical study of V2O5/HDA has been described. The X-ray diffraction data showed that the vanadium oxide presents a lamellar character as well as the presence of HDA into the layers indicating an intercalation reaction. The surface into the film promotes the formation of rods with diameter around 270 nm, and these rods are arranged almost parallel to each another. The cyclic voltammetry technique demonstrated that V2O5/HDA is highly stable after the 25th cycle, which occurs with little decrease in total charge during successive redox cycles. V2O5/HDA as a membrane on a pH-EGFET sensor demonstrated a linear behavior and a sensitivity of 38.1 mV/pH for the pH range 2–12. This value suggests that the material might need an increase in the number of actives sites, which can make vanadium oxide a potential component of an optimum device in the future.
References
Bergveld P (1970) IEEE Trans Biomed Eng BME 17:70
Bousse L, van den Vlekkert HH, de Rooij NF (1990) Sens Actuators B 2:103
Kreider KG, Tarlov MJ, Cline JP (1995) Sens Actuators B 28:167
Kwon DH, Cho BW, Kim CS, Sohn BK (1996) Sens Actuators B 34:441
Bergveld P (1974) IEEE Trans Biomed Eng BME 21:485
Chi LL, Chou JC, Chung WY, Sun TP, Hsiung SK (2000) Mater ChemPhys 63:19
Chang KM, Chao KY, Chou TW, Chang CT (2007) Jap J App Phys 46:4333
Chou JC, Chang JL, Wu CL (2005) Jap J App Phys 44:4838
Van der Speigel J, Lauks I, Chan P, Bablic D (1983) Sens Actuat B 4:291
Chou JC, Tzeng DJ (2006) Rare Metal Mat Eng 35:256
Liao YH, Chou JC (2006) Rare Metal Mat Eng 35:225
Batista PD, Mulato M, Graeff CFD, Fernandez FJR, Marques FD (2006) Bras J Phys 36:478
Batista PD, Mulato M (2005) App Phys Lett 87:143508
Guerra EM, Silva GR, Mulato M (2009) Solid State Sci 11:456
Nordlinder S, Nyholm L, Gustafsson T, Edström K (2006) Chem Mater 18:495
Tokudome H, Miyauchi M (2005) Angew Chem Int 44:1974
Oliveira HP, Graeff CFO, Brunello CA, Guerra EM (2000) J Non-Crystalline Solids 273:193
Guerra EM, Cestarolli DT, Da Silva LM, Oliveira HP (2009) J Solid State Electrochem. doi:10.1007/s10008-009-0877-3
Lira-Cantu M, Gomez-Romero P (1999) J Electrochem Soc 146:2029
Huguenin F, Torresi RM, Buttry DA (2002) J Electrochem Soc 149:A546
Guerra EM, Ciuffi KJ, Oliveira HP (2006) J Solid State Chem 179:3814
Guerra EM, Brunello CA, Graeff CFO, Oliveira HP (2002) J Solid State Chem 168:134
Fomichev VV, Vkrainskaya PI, Ilyin TM (1997) Spectrochim Acta Part A 53:1833
Dwyer CO, Lavayen V, Newcomb SB, Santa Ana MA, Benavente E, González G, Torres CMS (2007) J Electrochem Soc 154:K29
Chandrappa GT, Steunou N, Cassaignon S, Bauvais C, Livage J (2003) Catal Today 78:85
Dobley A, Ngala K, Yang S, Zavalij PY, Whittingham MS (2001) Chem Mater 13:4382
Malta M, Louarn G, Errien N, Torresi RM (2006) J Power Sources 156:533
Spahr ME, Stoschitzki-Bitterli P, Nesper N, Haas O, Novák O (1999) J Electrochem Soc 146:2780
Schöning MJ, Simonis A, Ruge C, Ecken H, Müller-Veggian M, Lüth H (2002) Sensors 2:11
Acknowledgments
This work was supported by FAPESP, CNPq and CAPES. We thank Prof. Herenilton P. Oliveira for allocation of his laboratory resources.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guerra, E.M., Mulato, M. Synthesis and characterization of vanadium oxide/hexadecylamine membrane and its application as pH-EGFET sensor. J Sol-Gel Sci Technol 52, 315–320 (2009). https://doi.org/10.1007/s10971-009-2062-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-009-2062-7