
Abstract
Liquid-state 29Si NMR was used to investigate the hydrolysis and condensation kinetics of ammonia-catalyzed tetraethoxysilane (TEOS) in methanol system. The reactive rate constants were calculated by applying first-order reaction approximation and the steady state approximation theory. The reaction orders with respect to TEOS, ammonia and water were derived, as well as the activation energies and the Arrhenius constants. It was found that the formation of intermediate species Si(OH)(OEt)3 was the rate-limiting step and its reaction rate equation was r TEOS=7.41×10−3[TEOS][NH3]0.333[H2O]0.227. Higher reactive temperature benefited the hydrolysis of TEOS. The results presented here indicated quantificationally that the formation of colloidal SiO2 particles was controlled by the initial hydrolysis of TEOS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Monodisperse silica nanoparticles have been widely applied in the fields of optics, electronics, catalysis, sensor and biology [1]. Stöber synthesis [2] was the first method to produce this kind of uniform silica nanoparticles through ammonia-catalyzed hydrolysis and condensation of ethoxysilanes in low molecule-weight alcohol. Thus-obtained spheric SiO2 particles may have diameters ranged from 5 nm to 2000 nm. The understanding on the kinetic of Stöber reaction is very helpful for the control of size and growth of SiO2 particles. Deep and detailed kinetics of Stöber synthesis were not obtained by the previous researches [3–8], and there were some contradiction between different explanations. And the exact reaction orders haven't been derived till now. In detail, there were two different viewpoints about the rate-limiting step of Stöber reaction to form SiO2 nucleus: the condensation of hydrolyzed monomer or the initial hydrolysis of TEOS. The work of Sadasivan [8] supported the first viewpoint, thinking a fully hydrolyzed dimmer (HO)3SiOSi(OH)3 was the key intermediate species. However, the second viewpoint was supported by Lee [3], Matsoukas [4], and Green [7] using light scattering, Raman spectroscopy, and liquid 29Si NMR technologies. Out of above considerations, the present work aimed to a deep understanding of the hydrolysis and condensation kinetics of TEOS under ammonia-catalysis and then to determine what should be the true rate-limiting step of Stöber reaction. To our knowledge, the precise rate constants and reaction orders obtained here was the first time.
2 Experiment
Reagent grade ammonium hydroxide (26% NH3), methanol, deionized and distilled water, and TEOS were used, and the preparation conditions of samples are presented in Table 1.


The typical 29Si NMR spectra of the solutions with ratio of TEOS:CH3OH:H2O:NH3 at (a) 1:12.5:4:0.18 (25°C), (c) 1:12.5:4:0.18 (45°C), (d) 1/12.5/4/0.045 (25°C), (h) 1/12.5/8/0.18 (25°C) (Continued on next page)
NMR sample was prepared by mixing two parts: solution A, TEOS was dissolved in half of the total methanol and stirred for 20 min; solution B, deionized water and aqueous ammonia hydroxide solution was dissolved into another half of the total methanol and stirred for 5 min. The reaction was initiated by mixing solution A and B. After vigorous stirring of 5 min, the mixture of solutions A and B was transferred to an NMR sample tube (5 mm O. D.) and immediately analyzed. Chromium (III) acetyl acetonate (Cr(acac)3, 1%) was added as the spin relaxation agent. Many studies [9, 10] have shown that Cr(acac)3 had no effect on the studied reactions. All 29Si NMR experiments were carried out in duplicate on a UNITY INOVA-500 Spectroscopy at a spectral frequency of 99.351 MHz. To achieve sufficient signal intensity, 168 scans were acquired for each spectrum with a 3 s pulse delay using a 90°pulse angle. During the experiments, gelation did not occur and the transesterification reaction can be ignored [7].
3 Results and discussion
3.1 \(^{{\rm 29}} {\rm Si}\) NMR spectra and data analysis
Liquid \(^{{\rm 29}} {\rm Si}\) NMR technology provides a very useful tool to obtain valuable information about the chemistry structures of silicon alkoxides and their hydrolyzed or condensed derivates [11, 12]. It allows identification and quantification of soluble Si-containing species in solution, thus enable kinetic data to be extracted. The disappearance of monomer, TEOS, from its initial level and the appearance of the intermediate species are presented by the typical 29Si NMR spectra (samples a, c, d, and h) in Fig. 1. In order to assign the 29Si NMR chemical shift for different soluble silicon species, the traditional notations were adapted [13]: Q presents the tetrafunctional silicon in TEOS. The symbol \({\it Q}_m^n\) denotes the products of hydrolysis or condensation of TEOS, where m and n are the number of Si–O–Si bridges and the number of silanol groups surrounding Si atom respectively. The detailed chemical shift of each soluble Si species and the corresponding molecular structure are listed in Table 2. According to the studies of Lee [3] and Green [7], the chemical shift of TEOS is at –81.3 ppm; the resonance signals of two hydrolysis intermediate species, \({\it Q}_{\rm 0}^{\rm 1}\) and \({\it Q}_{\rm 0}^{\rm 3}\), appear at –80.4 ppm and –78.3 ppm respectively; the NMR peak at –88.3 ppm corresponds to the dimmer \({\it Q}_1 ^0\). The resonance of \({\it Q}_0 ^2\) is not observed probably because of the fast condensation of the intermediate \({\it Q}_0 ^2\) under basic conditions [7].
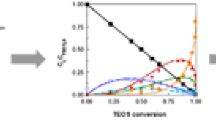
The relative concentrations of soluble intermediate Si species were determined by integrating the resonance peak in NMR spectra. The integrated area of the initial \({\it Q}_0 ^0\) peak of TEOS before the addition of catalyst and water was taken as 100%. Shown in Fig. 2, the typical time-dependent concentration (samples a, c, d, and h) is the base of further calculation of the rate constants. In Fig. 2, the time-dependence of monomer concentration (\({\it Q}_0 ^0\)) was obtained by fitting the experimental data with an exponential decay of first order. The time-dependences of the intermediate species concentrations (\({\it Q}_0 ^1\), \({\it Q}_0 ^3\), \({\it Q}_1 ^0\)) were obtained only to guide the eye. The effect of reaction temperature on the kinetics is obtained by comparing samples (a), (b) and (c). The effect of ammonia content on the kinetics is obtained by comparing samples (a), (d) and (e). The effect of water content on the kinetics is obtained by comparing samples (a), (f), (g) and (h). Therefore a qualitative conclusion can be easily drawn from Figs. 1 and 2: the hydrolysis of TEOS can be speeded up by increasing the ammonia content, water content, or reactive temperature.

The time-dependent concentrations of soluble Si species for the solutions with ratio of TEOS:CH3OH:H2O:NH3 at (a) 1:12.5:4:0.18 (25°C), (c) 1:12.5:4:0.18 (45°C), (d) 1/12.5/4/0.045 (25°C), (h) 1/12.5/8/0.18 (25°C). ■—\({\it Q}_0 ^3\); ●—\({\it Q}_0 ^1\); ▲—\({\it Q}_0 ^0\); ▼—\({\it Q}_1 ^0\)
3.2 Reaction kinetics of Stöber synthesis
The overall hydrolysis of TEOS can be described as equation:
In fact, the hydrolysis of precursor is a step-by-step process and it runs nearly simultaneously with the condensation that makes the reaction mechanism more complex. So based on this consideration, Harris [6] and Lee [3] suggested a detailed reaction model and a simple reaction model respectively. In fact it is impossible to calculate the exact rate constants according to the detailed model. So a simple hydrolysis and condensation model of TEOS is adopted here like follow according to Green's proposal [7].
where k h1, k h2, and k c are the rate constants for Eqs. (2)–(4), respectively. In fact, \({\it Q}_0 ^3\) species can condense with each other to form \({\it Q}_1 ^4\) like Eq. (4), but \({\it Q}_1 ^4 \) can't be detected in experiments, so this reaction isn't taken into account here. Considering that the condensation species can be easily detected even for those species larger than \({\it Q}_1 ^4 \), the invisibility of \({\it Q}_1 ^4 \) may be due to the very low concentration of it in tested solution. In this case, there must be a fast exhaustion of \({\it Q}_1 ^4 \) that make its concentration lower than the NMR detective limit. The most possible reason for this exhaustion is because \({\it Q}_1 ^4 \) quickly participate in the further nucleation and disappear in the “eye” of NMR instrument.

The time-dependences of TEOS concentration with the molar ratio of TEOS:CH3OH:H2O:NH3 at (a) 1:12.5:4:0.18 (25°C), (b) 1:12.5:4:0.18 (35°C), (c) 1:12.5:4:0.18 (45°C), (d) 1/12.5/4/0.045 (25°C), (e) 1/12.5/4/0.36 (25°C), (f) 1/12.5/2/0.18 (25°C), (g) 1/12.5/6/0.18 (25°C), (h) 1/12.5/8/0.18 (25°C). The slop of the straight line is the pseudo first-order rate constant \(k_{\rm h}^{\prime}\) of the hydrolysis reaction
To obtain the reaction orders of the hydrolysis and condensation of TEOS, the concentrations of the starting reactants were varied. The rate equation for the reaction (2) is as follows
where the exponents α, β and γ are the reaction orders belonging to TEOS, NH3 and H2O respectively. Since NH3 is used as the catalyst during the hydrolysis and the condensation, its concentration can be considered constant. The change of water content was less than 5% of the initial level [14], so it is also considered invariable. If we define
the rate Eq. (5) can be simplified to
As can be seen in Fig. 2, the exponentially fitted time-dependent concentration of \({\it Q}_0^0 \) is a good approximation for the experimental data. In this case, it is assumed that the reaction (2) is of first-order, i.e., α=1, so the integration of Eq. (7) leads to
Thus a plot of ln[\({\it Q}_0 ^0\)] vs. t should yield a straight line with \(-k_{\rm h}^{\prime} \) as slope and \({\rm ln}[{\it Q}_0 ^0]_0\) as intercept, which are shown in Fig. 3. This good linearity proves α=1 indeed. Furthermore, taking the logarithm of Eq. (6) will give
Thus a linear plot of \({\rm ln}k_{\rm h}^{\prime}\) vs. ln[NH3] will produce the reaction order β of NH3 when [H2O] remains constant. This plot is shown in Fig. 4. Similarly, a linear plot of \({\rm ln}k_{\rm h}^{\prime}\) vs. ln[H2O], shown in Fig. 5, produces the reaction order γ of H2O when [NH3] remains constant. Directly from Figs. 4 and 5, β=0.333 and γ=0.227 have been calculated. Applying \({\rm ln}k_{\rm h}^{\prime}\) obtained from Fig. 3, β and γ in Eq. (6), k h1 has been calculated and listed in Table 3. Finally at 25°C the initial hydrolysis rate equation of TEOS is

The pseudo first-order rate constant \( k_{\rm h}^{\prime} \) vs. ammonium concentration for the solutions with molar ratio of TEOS:CH3OH:H2O:NH3 at (a) 1:12.5:4:0.18 (25°C), (d) 1/12.0/4/0.045 (25°C), (e) 1/12.5/4/0.36 (25°C)

The pseudo first-order rate constant \( k_{\rm h}^{\prime} \) vs. water concentration for the solutions with molar ratio of TEOS:CH3OH:H2O:NH3 at (a) 1:12.5:4:0.18 (25°C), (f) 1/12.5/2/0.18 (25°C), (g) 1/12.5/6/0.18 (25°C), (h) 1/12.5/8/0.18 (25°C)
From Eqs. (2) and (3), the disappearance of \({\it Q}_0 ^1\) is accompanied by the appearance of \({\it Q}_0 ^3\). But the intermediate \({\it Q}_0 ^1\) is so reactive that it has little time to accumulate. After \({\it Q}_0 ^1\) content approaches its maximum, it reaches a steady state. According to the steady-state approximation [15, 16], following equation has been acquired
where \([Q_0 ^0 ]_{\rm SS}\) and \([{Q_0 ^1} ]_{\rm SS}\) are the steady-state concentrations. Hence the second hydrolysis rate constant k h2 can be defined by
The obtained k h2 are also listed in Table 3. From Eqs. (3) and (4), the rate constant k c is calculated when applying the steady-state approximation to \({\it Q}_0 ^3\). The results of k c are also summarized in Table 3.
The 29Si NMR spectra of the solutions reacting at 25°C (a) and 45°C (c), are shown in Fig. 1. The temperature-dependence of rate constants can be represented by the Arrhenius equation,

where k is the rate constants of reactions (2)–(4); A is the Arrhenius factor; E a is the activation energy of reaction (2)–(4); T is the temperature and R is the ideal gas constant (8.3145 J·mol-1·K-1). The plot of lnk vs. 1/T is shown in Fig. 6 whose slope gives –E a/R, and the intercept is lnA. So reaction activation energies and Arrhenius factors of reaction (2)–(4) are summarized in Table 4.

Arrhenius plots of solution with TEOS:CH3OH:H2O:NH3 at 1:12.5:4:0.18
3.3 Determination of rate-limiting step
Seen in Table 3, k h1 is one order less than k h2, and k h2 is one order less than k c. In another word, the reaction rate is gradually increased along a sequence of k h1<k h2<k c, which can be explained by S N 2-Si nucleophilic replacement mechanism [17, 18]. Under basic conditions, it is likely that water dissociates to produce nucleophilic hydroxyl anions in a rapid step and then the hydroxyl anion attacks the silicon atom. Iler [19] and Keefer [20] proposed an S N 2-Si mechanism in which OH‒ displaces EtO‒ with inversion of the silicon tetrahedron.
With monomer's hydrolysis proceeding, ‒OEt is gradually substituted for ‒OH. Because the electron-withdrawing capabilities of ‒OH is stronger than that of ‒OEt, the replacement of ‒OEt by ‒OH leads to more positive charge on Si atom, which benefits the further nucleophilic attack of OH‒ to Si+. So the subsequent hydrolysis occurs more quickly. At the same time, it is common that the condensation of ethoxysilanes is faster than the hydrolysis under basic conditions. From the above-mentioned standpoints, reaction (2) is the slowest one in the continuous hydrolysis and condensation, indicating that the first hydrolysis of TEOS is the rate-limiting step and the formed \({\it Q}_{\rm 0}^{\rm 1}\) is the key intermediate species. Our results proved quantitatively that the initial hydrolysis of TEOS was the rate-limiting step, which was consistent with the results of Lee [3], Matsoukas [4] and Green [7], but contradicted with the viewpoints of references [5, 21].
From Fig. 1 and Table 2, no resonance signal of \({\it Q}_{\rm 0}^{\rm 2}\) was detected during the evolving period of TEOS. In fact, it is very difficult to observe this signal because of the fast consumption of \({\it Q}_{\rm 0}^{\rm 2}\). According to Green's study [7], the NMR signal of \({\it Q}_0 ^2\) can be detected only when the concentrations of water and ammonia are very low because in this case the total reaction rate is slow and the consumption of \({\it Q}_0 ^2\) is slowed down too. Therefore intermediate \({\it Q}_0 ^2\) is only a short-life transition species that will converse to \({\it Q}_0 ^3\) at once. In addition, the concentration sum of all the detectable Si species except for TEOS is 1%–8% of the total Si content in composition. The disappeared Si source mainly participates nucleation and becomes insoluble and undetectable by liquid NMR.
From Table 4, the activation energy increases from reaction (2), (3) to (4). The activation energy of reaction (2) is the lowest in those tree reactions, indicating that reaction (2) is the easiest to occur. But simultaneously reaction (2) is the slowest in those tree reactions, which seems to contradict with its activation energy. If the Arrhenius factors are taken into account, the contribution of Arrhenius factor to rate constant should be much more than that of activation energy. So reaction (2) becomes the rate-limiting step although its activation energy is less than those two reactions.
3.4 Comparison with the previous researches
From 1990s then on, many efforts have been made to correctly understand the reaction mechanism of the sol-gel process of metal alkoxides among which organic siloxane may be the simplest sol-gel precursor. However, this is not an easy work to track the true sol-gel process even its initial stage because the formation of so-called sol involves complex chemical reactions and a variation from homogeneous solution of precursor to heterogeneous colloidal suspension of particles. In the previous researches, several technologies have been employed to investigate the reaction mechanism and the subsequent particle growth mechanism, including nuclear magnetic resonance, Raman spectroscopy, gas chromatography, conductivity, infrared spectroscopy, dynamic light scattering, and small angle X-ray scattering. Although much progress has been made in this field, the precise chemical reaction kinetics is still a problem.
Reported in 1990, considering ammonia as the base catalyst and the ionization reaction of ammonia with water, Harris [6] deduced a reaction rate equation like follow:
The rate Eq. (15) is based on the overall hydrolysis and condensation reactions as follows:
However the hydrolysis of TEOS is a step-by-step process and runs nearly simultaneously with the condensation. Therefore Eq. (16) is only an ideal expression to the true reaction situation. The reaction mechanism should be more complex. In fact, the hydrolysis rate constant obtained by Harris, \( k_{\rm H}^\ast \), was a collective reflection of hydrolysis rates of all steps, not same as the k h1 obtained by us. Although Eq. (15) takes the same form as Eq. (10), the obviously different exponentials reflect the entirely different calculating base. Additionally, the condensation rate constant obtained by Harris, \( k_{\rm C}^\ast \), was taken by assuming that the hydrolysis and condensation of TEOS can be completely separated like Eqs. (16) and (17). So \(k_{\rm H}^\ast \) and \(k_{\rm C}^\ast \) were not obtained on the same level of mechanism as k h1, k h2, and k c in this paper.
In 1997, Lee and coworker [3] reported an in-situ investigation of Stöber reaction by 29Si NMR, photon correlation spectroscopy, and conductivity. They thought that, since in an ordinary Stöber system there is only one reaction intermediate detectable by 29Si NMR, an acid-base two-step catalysis to TEOS should be employed. Thus, there were eight NMR signals could be detected. But this experiment method did not reflect the true condition of Stöber reaction either. Therefore, the two-step procedure, especially employed in Lee's study, makes their results not directly comparable with the results presented in this paper. Although, they obtained a valuable conclusion that the nucleation of Stöber reaction was rate-limited by the hydrolysis of the singly hydrolyzed monomer.
The most important experimental results about this study were reported by Green and his coworker [7] in 2003. In this literature, they first confirmed that transesterification between methanol and TEOS did occur, but it was negligible compared to the production of hydrolyzed intermediates. This conclusion ensured the reliability of in-situ 29Si NMR experimental results. Another important contribution was that they proved the \({\it Q}_1 ^6\) was not the hydrolysis intermediate of TEOS. Indeed, intermediate \({\it Q}_1 ^6\) wasn't detected in our all experiments either. If possible, \({\it Q}_1 ^6\) should be the condensation product of full-hydrolyzed species Si(OH)4, but Si(OH)4 is unstable in Stöber system due to the fast condensation of Si-OH groups. Therefore, it is reasonable that \({\it Q}_1 ^6\) doesn't exist. In addition, concerning the effects of water and ammonia concentrations, they used 29Si NMR to investigate the ammonia-catalyzed hydrolysis of TEOS in methanol and ethanol ([TEOS] = 0.5 M; [NH3] = 0.05–0.1 M; and [H2O] = 1.1–4.4 M). Their research showed that the rate constant of the rate-limiting step was largely dependent on [H2O] and [NH3]. However, a true rate constant shouldn't be dependent on the concentrations of any reactants or catalyst. In fact, their study dealt with four 29Si NMR experiments in which [H2O] and [NH3] were set three levels respectively. Based on such few experiments, it was difficult to disclose the precise reaction rate constants. In our experiment design, all the conditions needed for extracting kinetics parameters have been taken into account. The data fluctuation of our obtained reaction rate constants was within an acceptable error range. So it is the first time to obtain the precise kinetics parameters of Stöber reaction. Based on our experimental results, the key hydrolysis intermediate, \({\it Q}_0 ^1\), was quantitatively confirmed as well as the rate-limiting step, which was consistent with the conclusion of Green [7].
4 Conclusions
The ammonia-catalyzed hydrolysis and condensation of TEOS in methanol were investigated by in-situ liquid 29Si NMR. The growth of colloidal particles was quantificationally clarified to be controlled by the initial hydrolysis of TEOS. For the first time, a whole set of rate constants of both the rate-limiting step and the non-rate-limiting steps were obtained. Besides, the reactive orders, activation energy, and the Arrhenius factors were calculated. This study proved that the rate-limiting step is the first-step hydrolysis of TEOS and \({\it Q}_0 ^1\) is the key intermediate species for the whole reaction process.
References
Shingo K, Ikuko Y, Noriko Y (1997) In: Dunn BS, Mackenzzie JD, Pope EJ, Schmidt HK, Yanane M (eds) Sol-Gel Optics IV, SPIE vol. 3136. The international society for optical engineering, Bellingham, Washington
Stöber W, Fink A, Bohn E (1968) J Colloid Interface Sci 26:62
Lee K, Look JL, Harris MT, McCormick AV (1997) J Colloid Interf Sci 194:78
Matsoukas T, Gulari E (1988) J Colloid Interface Sci 124:252
Bogush GH, Zukoski CF (1991) J Colloid Interface Sci 142:1
Harris MT, Brunson RR, Byers CH (1990) J Non-Cryst Solids 121:397
Green DL, Jayasundara S, Yui-Fai Lam, Harris MT (2003) J Non-Cryst Solids 315:166
Sadasivan S, Duber AK, Li Y, Rasmussen DH (1998) J Sol-Gel Sci Tech 5:12
Harris RK, Kimber BJ (1974) J Organometal Chem 70:43
Bailey JK, McCartney ML (1992) Colloids Surf 151:63
Assink RA, Kay BD (1991) Ann Rev Mater Sci 21:491
Sugahara Y, Okada S, Kuroda K, Kato C (1992) J Non-Cryst Solids 139:25
Brinker CJ, Scherer GW (1990) Sol-gel science: the physics and chemistry of sol-gel processing. Academic Press, San Diego
Fyfe CA, Aroca PP (1997) J Phys Chem B 101:9504
Volk L, Richardson W (1977) J Chem Educ 54:95
David Cater E (1983) J Chem Educ 60:109
Parker AJ (1967) Adv Phys Org Chem 5:173
Parker AJ (1969) Chem Rev 1:69
Iler RK (1979) The chemistry of silica. Wiley, New York
Keefer KD (1984) In: Brinker CJ, Clark DE Ulrich DR (eds) Better ceramics through chemistry. North-Holland, New York, p 15
Boukari H, Long GG, Harris MT (2000) J Colloid Interf Sci 229:129
Acknowledgments
The financial support from the National Key Native Science Foundation (No. 20133040) was gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, Y., Wu, D., Sun, Y. et al. A new study on the kinetics of Stöber synthesis by in-situ liquid 29Si NMR. J Sol-Gel Sci Technol 42, 13–20 (2007). https://doi.org/10.1007/s10971-006-1518-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-006-1518-2