
Abstract
High-resolution gamma-spectrometry and ICP-MS measurements were utilized to confirm the validity of secular equilibrium among the identified natural radioactive progeny of 238U and 232Th series. The measurements of 238U, 232Th, and 40K concentrations, in the soil within 2 km range around the first nuclear reactor in Jordan, were close to the worldwide average levels. Among artificial radionuclides, only 137Cs was detected but with very low traces. The dose rate and radiological hazard parameters were found to be close to worldwide average values and below the recommended limits. Our results indicate that secular equilibrium is unperturbed within and around the uncontaminated reactor site.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Jordan Research and Training Reactor (JRTR) is the first facility capable of producing a sustained nuclear chain reaction in Jordan. The facility has been constructed on the campus of Jordan University of Science and Technology (JUST) that is located in Irbid governorate; the largest governorate in northern Jordan. The reactor is operated by Jordan Atomic Energy Commission (JAEC) and it had reached its first criticality, after nuclear fuel installment, in April 2016. JRTR is a 5MWth multi-purpose research reactor that can hold enough critical mass to sustain a nuclear chain reaction. This research reactor will be used to build a local capacity in the fields of nuclear engineering, nuclear reactor operations, forensic analysis, radioisotope production, and neutron beam applications. The establishment of this facility comes as a preparatory work for the national plan to prepare qualified cadres and gain sufficient knowledge before embarking the run of the planned national scale nuclear power plant [1, 2].
The International Atomic Energy Agency (IAEA) Code of Conduct on the Safety of Research Reactors monitors research reactors safety issues. The code entails a complete and systematic procedure for a safe nuclear reactor lifetime operation. This is get accomplished via peer review missions to assess and evaluate the safety related to the environmental and health aspects within the reactor influence zone [3]. To perform this, certain periodic environmental studies are applied in the reactor zone before, during, and after the reactor operation. One of the most important aspects is a pre-commissioning radiological assessment of the reactor site before reactor operation. In such study, the direct radiation exposure assessment can be extracted directly from the measurements of radionuclide concentrations in soil [4]. These measurements are essential as they serve as a baseline for the future studies of the reactor radiological impact on its surrounding environment during and after reactor operation [5, 6].
Among the natural radionuclides, uranium (238U), Thorium (232Th) and Potassium (40K) are considered the most important for identifying radioactivity levels due to their common presence at varied levels in all soil types [4]. Along with their series of radioactive decay chains, 238U and 232Th, produce radioactive daughters that induce radiation exposures [7,8,9,10]. Soil could be rich with these radionuclides with various ratios depending primarily on the geological conditions [7]. Radionuclides within the soil are easily transferred to plants and to the rest of the food chain. In addition, most building materials are made up of soil and rocks and the materials-air interference plays a vital part in the human exposure [8, 11, 12]. The natural radioactivity can vary greatly from one location to the other but radioactivity due to terrestrial radionuclides is considered a main contributor to the total dose received due to external radiation exposure by living organisms [4].
In the literature, typically, natural radionuclides concentrations are measured in soil using gamma spectrometry (γ-spectrometry) assuming secular equilibrium. However, secular equilibrium can be perturbed due to radionuclides loss or gain within the decay series of 238U and 232Th. These changes in the isotopic system can take place by physical or geochemical processes such as recrystallization and water–rock interaction affecting the more geochemically soluble radionuclides [13, 14]. In this work, high-resolution γ-spectrometry was adopted to detect and measure natural and artificial radionuclides’ concentrations in soil samples that were collected from locations inside and surrounding JRTR campus prior to nuclear fuel arrival to the reactor site. In addition, inductively coupled plasma-mass spectrometry (ICP-MS) method was used to investigate the validity of secular equilibrium among the natural radionuclides and confirm their concentration levels measured by the high-resolution γ-spectrometry technique. The obtained measurements will be used as baseline data for the future studies tracking the impact of reactor operation on radioactivity levels within the reactor zone and its nearby surrounding area. Furthermore, the current results are used to evaluate the radiological hazards due to the identified radionuclides concentrations.
Experimental
Samples’ collection and physical treatment
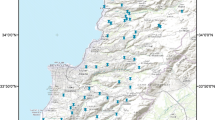
Soil samples were collected from nineteen locations inside JRTR campus and areas surrounding the reactor building, during October 2015, prior to JRTR nuclear fuel loading and hot commissioning phase. The samples were collected at two varied distances from the reactor core building as a center viz. 500 m and 2 km as shown in Fig. 1. The coordinates of locations were determined using a global positioning system. Only two of the sampling locations (B6 and B8) were unreachable due to construction activities. The other 17 samples were collected from uncultivated surfaces of soil using a custom-made tool according to the IAEA sampling procedure [15]. The collected samples were mixed and cleaned from stones, pebbles, leaves, and roots and dried in the oven at 104 °C for 24 h. After cooling down, a jaw crusher was used to smash the samples. To convert samples into fine powder and reduce the size of particles, they were further grinded using a disk mill. Samples were then ball-milled, sieved using 60 μm mesh sieves. To avoid contamination, the machines were cleaned before treating each of the samples.

A map showing soil sampling locations inside and around JRTR site
γ-spectrometry
After the sieving step, samples were weighed and packed in plastic cylinders of 7.5 cm diameter and 1.5 cm height. These containers were tightly sealed, and stored for 2 months before counting for achieving secular equilibrium between radium and its progeny [11]. The samples’ gamma spectra were collected using an ORTEC high-purity germanium (HPGe) detector (GEM50-83 model) with 50% relative efficiency and 0.8 keV Full-Width Half Maximum (FWHM) at 122 keV gamma line of 57Co and 1.9 keV FWHM at 1332.5 keV gamma line of 60Co. The detector is coupled with 16 k channel multichannel analyzer (MCA) and enclosed with lead, aluminum and copper shields to reduce signal noise due to ambient background radiation. The efficiency and energy calibration curves were derived using standard sources of type CBSS 2 provided by Czech Metrology Institute (CMI) that cover the energy range from 59.5 keV to 1.836 MeV. To reduce statistical uncertainty; the counting time was 86,400 s. The resulted gamma-ray spectrum peaks, when are unveiled in sufficient quantities in the test sample, give an indication of the nature of the existing radionuclides. If multiple peaks give an indication for the same radioisotope, then a weighted mean activity is calculated [10]. Some energy peaks may infer more than one radionuclide or are of low yields, such energy peaks, are excluded from measuring that radionuclide concentration level.
The specific activity, A, of the radionuclides present in a sample is evaluated at each energy photopeak using the following relation [9]:
where C net is the corrected net peak count (background counts are subtracted), ɛ is the full energy peak efficiency, P γ is the probability of gamma-ray emission, t is the live time of detector in seconds, m is the dry-mass of the measured sample in kg. This equation is used for determining the activity using a single energy photopeak of a certain radioisotope. If more than one peak is identified for the radioisotope, the peaks activities are averaged [10]. Assuming secular equilibrium condition, 238U (226Ra) activity is calculated based on the arithmetic mean of 214Bi (609.3 keV) and214Pb (295.2 and 351.92 keV) activities, while, 232Th is evaluated as the average of the available activities of 212Pb (238.6 keV), 208Tl (583.2 keV), and 228Ac (911.2 keV) [10]. On the other hand, 40K and 137Cs were calculated based on their prominent gamma lines at 1460.8 and 661.7 keV, respectively [12]. Minimum Detectable Activity (MDA) and uncertainty calculations were calculated as described elsewhere [16]. Any radionuclide with activity level below the MDA was excluded from the average activity calculations.
ICP-MS
Samples chemical treatment for ICP-MS
To detect uranium and thorium concentrations using ICP-MS, the samples must be treated physically and then chemically. 0.25 g of each physically pretreated sample, as described above, was taken using a calibrated scale. The samples were, chemically, digested by adding ultra-high-purity grade acids; 3 mL of concentrated nitric acid (HNO3; 70%) and then 2 mL of Hydrogen peroxide (H2O2; 30%) and, to enhance the digestion step, samples were heated to 70 °C using a Digiblock overnight for organic materials removal [17]. Then, the digested samples were left to be dried overnight using the Digiblock at constant temperature at 200 °C to assure full dryness. Consequently, 15 mL of 25% concentrated HNO3 was added to dissolve the samples, heated at 150 °C, and left for 30 min to ensure that the sample is totally dissolved in the acid. After chemical digestion, the samples were diluted by mixing with water of high purity for reaching a 25 mL solution. 20 µL of this solution was then added to nitric acid of 2% concentration to have a dilution factor of 500 by making a 10 mL sample.
ICP-MS analysis
Prior to the analysis, six standard sources with varied known concentrations were used to derive the linear intensity versus concentration calibration curve. Whenever any modification in the ICP-MS instrument (BRUKER 820-MS) settings occurs, a mixed standard source, from AccuStandard consisting of nine elements of the 5 ppm concentration of varied atomic weights, is used to check the ICP-MS optimization. The ICP-MS optimization resulted in 0.8 amu resolution. Four types of samples were used for the ICP-MS analysis, a blank sample, a Certified Reference Materials sample (CRM), the samples of interest, and replicates of the samples of interest, all were analyzed separately with an identical procedure. A calibration blank is a zero standard for instrument calibration and is used with every batch of samples to detect any contamination occurred during the measurement process. The main objective of CRMs is to validate the whole process, the chemical digestion, and ICP-MS analysis, in terms of recoveries besides the uncertainty calculation. Three replicates of each sample were prepared and analyzed. The concentration, CN, was corrected against dilution using the following relation:
where CN b is the raw concentration obtained from the calibration curve, DF 1 is the dilution factor, which equals 500, DF 2 is the digestion dilution factor and equals 100 in this work. Each replicate was measured three times and an average value of the nine readings was obtained. The obtained ICP-MS results, in part per million (ppm) concentrations, were converted into specific activities (Bq kg−1); where 1 ppm of U makes 12.43 Bq kg−1 of specific activity and 1 ppm of Th is equivalent to 4.07 Bq kg−1 [5].
Evaluation of radiological hazards
In addition to the dose rates, the related radiation hazard and risk parameters are calculated as follows:
Absorbed dose rate in air
Using the measured activities, the absorbed dose rate in the air at 1 m above the ground surface, \(D\), can be calculated using the following relation [4, 6, 8]:
where A represents the specific activity in \({\text{Bq kg}}^{ - 1}\) and the subscripts describe the identified radionuclide, and \(C_{\text{Ra}} = 0.462; C_{\text{Th}} = 0.604;\) and C K = 0.0417 are the conversion factors for the dose rates.
Annual effective dose equivalent
The outdoor annual effective dose equivalent (AEDE), assuming 20% for outdoor occupancy factor received by the adults due to natural radioactivity in soil can be calculated using the following relation [7,8,9]:
where the conversion coefficient, CF, equals \(0.7 \, {\text{Sv}}/{\text{Gy}}\), and T is the annual time of exposure (T = 8760 h) and 0.2 is outdoor occupancy factor.
For the relations above and for the discussion below, it is assumed that all decay products of 238U and 232Th series are in equilibrium. Hence, the activity concentrations of 238U and 226Ra can be used interchangeably [9]. This assumption of radioactive equilibrium is truly valid as will be deliberated in the results and discussion section.
Radium equivalent activity
To describe the radiation hazards associated with a mixture of various radionuclides, radium equivalent activity, Ra eq, radiological hazard index has been introduced. This index gives a reasonable representation of hazard related to the commonly non-uniform distribution of natural radionuclides. The definition of this index implies that 100 Bq kg−1 of 226Ra, 70 Bq kg−1 of 232Th, or 1300 Bq kg−1 of 40K have equivalent dose due to gamma radiation [7]. The amount of Ra eq that is attributed to the presence of 226Ra (238U), 232Th, and 40K is calculated using the following relation [7, 8]:
The external and internal hazard indices
The external and the internal hazard indices are widely used [7, 8, 11]. While the external hazard index (H ex) deals with the external radiation effects, the internal hazard index (H in) is used for evaluating the radiation effects caused by radon (222Rn) and its progeny to the respiratory organs. These hazard indices can be evaluated using the following relations, respectively:
The external and the internal hazard indices should be less than unity; this requirement ensures safe use and negligible hazardous effects to the respiratory system [8].
Excess lifetime cancer risk
To assess the extra probability of developing cancer due to the radiation exposure effects during the lifespan of a person, the excess lifetime cancer risk, ELCR, has been calculated using the following relation [12]:
In this equation, LE is the life expectancy in Jordan that is 74 years, and RF is the mean fatal risk factor per Sievert that is 0.05 Sv−1.
Results and discussion
Secular equilibrium
The assumption of secular equilibrium was investigated, first, by calculating the ratios of the specific activities of all identified radionuclides using γ-spectrometry and, then, by comparing the γ-spectrometry results for uranium and thorium concentrations with those yielded from ICP-MS. As depicted in Fig. 2, using γ-spectrometry, the ratios of activities of all of the identified radionuclides, within both 238U and 232Th series, are close to unity.

Ratios of natural radionuclides’ activities within 238U and 232Th series identified by γ-spectrometry
In addition, there is a clear agreement among the results obtained for 238U and 232Th concentrations by γ-spectrometry and ICP-MS as illustrated in Fig. 3 that shows the correlation of the results of samples analyzed by both of the techniques. The correlations for both 238U and 232Th concentrations are in very good agreement with Pearson’s correlation coefficient of values higher than 0.95 with a close 1:1 correspondence. This indicates that all of the studied radionuclides are in secular equilibrium and confirms the calculated activities using γ-spectrometry.

Correlation of γ-spectrometry and ICP-MS concentrations for 238U and 232Th using results of 16 samples analyzed by both techniques
Activity concentrations of identified radionuclides
The gamma spectra of all of the samples were analyzed to deduce the concentration activities of all identified radionuclides that have gamma photopeaks with sufficient number of counts; those with specific activities that are higher than the corresponding MDAs. Only gamma lines related to 214Bi, and 214Pb (238U series); 212Pb, 208Tl, and 228Ac (232Th series); and 40K were evident in the spectra without interference. In addition, among artificial radionuclides, only low traces of 137Cs were present in most of the samples excluding one sample, C2, where it has not been detected. Figure 4 shows the calculated activity concentrations of the identified radionuclides in the soil samples collected from the JRTR campus and its close surrounding area as shown in Fig. 1.

Variation of identified radionuclides’ activity concentrations (238U, 232Th, 40K, and 137Cs) in the soil samples collected from areas inside and the surrounding proximity of JRTR campus. Solid lines represent the average activity concentration for each radionuclide
238U activity concentration ranged between 21.2 and 33.4 Bq kg−1 with an average value of 28.5 Bq kg−1, where all of the 238U concentration levels are lower than the world average of 35 Bq kg−1 [12, 18]. 232Th specific activity ranged between 27.7 and 42.9 Bq kg−1 with an average of 38.3 Bq kg−1, which is to some extent higher than the world average level of 30 Bq kg−1 [7, 18]. 40K specific activity ranged between 246.1 and 555.8 Bq kg−1 with an average value of 450.1 Bq kg−1, which is higher than the world average level of 400 Bq kg−1 [7, 18]. The slight increase in 40K activity can be attributed to the practice of mixing of the soil with fertilizers that are rich with 40K due to the relatively wide agriculture activity around JRTR campus. As depicted in Fig. 5, there is a trend of clear positive correlation among these radionuclides. In addition, Fig. 6 shows that the ratios of these radionuclides have to some extent reasonable relative standard deviations (RSD) around their means with values of 11.34%, 15.96%, and 17.66% for 238U/232Th, 232Th/40K, and 238U/40K, respectively. Furthermore, the activity measurements were found to be random in space with no explicit spatial coherence as could be deduced from Fig. 4. These findings, in addition to the positive correlation, may indicate that there are no substantial differences in the geochemical composition and origin of soil types within the study area [7, 12].

Correlations of the identified natural radionuclides: 238U, 232Th, and 40K. The dotted lines represent the coordinates for world average values for each of the identified radionuclides

Ratios of identified natural radionuclides: a 238U/232Th, b 232Th/40K, and c 238U/40K. The dotted lines represent the means for these ratios; 0.7472, 0.0874, and, 0.0651, respectively
Cs137 is an artificial radionuclide that spreads globally after nuclear accidents especially Chernobyl accident or nuclear weapon tests and its appearance within soil contents are limited by IAEA missions with a range between 5 and 100 Bq kg−1 [18]. All samples, showed that 137Cs values were much below this range, where the highest value was 6.7 Bq kg−1 for sample A7 with an average value of 3.9 Bq kg−1.
Radiological hazards assessment
The obtained activity concentrations due to the identified natural radionuclides 238U (226Ra),232Th, and 40K within the soil samples inside and around JRTR campus have been measured with suitable assumptions to derive the most representative measure of radiological hazards. Table 1 summarizes the range and mean values of calculated absorbed dose rate in air (D), annual effective dose equivalent (AEDE), radium equivalent (Ra eq), internal hazard index (H in), external hazard index (H ex), and excess lifetime cancer risk (ELCR) for the soil samples within the 2 km range of JRTR site.
While few samples have values of absorbed dose in air and annual effective dose equivalent that are slightly higher than those that represent the worldwide average values [5, 7], on the contrary, and for all of the studied samples, the radiation hazard parameters of Ra eq, H in, H ex, and ELCR were all below the recommended levels and worldwide average values for all of the samples [5, 7]. Thus, it can be inferred that there are no significant radiological hazards at JRTR site prior to loading of nuclear fuel and reactor operation.
Conclusions
The use of two nuclear analytical methods, viz. high-resolution γ-spectrometry and ICP-MS, indicates that secular equilibrium is well achieved among 238U and 232Th series in the analyzed soil samples. The concentrations of natural radionuclides 238U, 232Th, 40K, within 2 km range of JRTR site were found to be close to the worldwide average values. Among the artificial radionuclides and fission products, only 137Cs was detected but with very low values compared to the worldwide range. The calculated radiological hazard parameters were close to the worldwide average values or below the permissible limits. Our results show that the site of the first nuclear reactor in Jordan, before nuclear fuel loading and reactor operation, is an uncontaminated area with no abnormal radioactivity levels. In addition, the current results should form the baseline data that is required for any future radiological assessment of JRTR site during and after its operational phase.
References
Jordan’s First Nuclear Research Reactor Goes Through IAEA Peer Review, IAEA Office of Public Information and Communication. https://www.iaea.org/newscenter/news/jordans-first-nuclear-research-reactor-goes-through-iaea-peer-review. Accessed 27 Jun 2017
JAEC (2011) White paper on nuclear energy in Jordan. Jordan Atomic Energy Commission, Amman
IAEA (2016) Safety of research reactors. IAEA safety standards series. International Atomic Energy Agency, Vienna
UNSCEAR (2008) Sources and effects of ionizing radiation. United Nations Publications, New York
Kritsananuwat R, Sahoo SK, Fukushi M, Pangza K, Chanyotha S (2015) Radiological risk assessment of 238U, 232Th and 40K in Thailand coastal sediments at selected areas proposed for nuclear power plant sites. J Radioanal Nucl Chem 303(1):325–334. doi:10.1007/s10967-014-3376-7
Veiga R, Sanches N, Anjos RM, Macario K, Bastos J, Iguatemy M, Aguiar JG, Santos AMA, Mosquera B, Carvalho C, Baptista Filho M, Umisedo NK (2006) Measurement of natural radioactivity in Brazilian beach sands. Radiat Meas 41(2):189–196. doi:10.1016/j.radmeas.2005.05.001
Tabar E, Yakut H, Saç MM, Taşköprü C, İçhedef M, Kuş A (2017) Natural radioactivity levels and related risk assessment in soil samples from Sakarya, Turkey. J Radioanal Nucl Chem. doi:10.1007/s10967-017-5266-2
Singh P, Singh P, Bajwa BS, Sahoo BK (2017) Radionuclide contents and their correlation with radon-thoron exhalation in soil samples from mineralized zone of Himachal Pradesh. India. J Radioanal Nucl Chem 311(1):253–261. doi:10.1007/s10967-016-4975-2
Pehlivanovic B, Avdic S, Gazdic I, Osmanovic A (2017) Measurement of natural environmental radioactivity and estimation of population exposure in Bihac, Bosnia and Herzegovina. J Radioanal Nucl Chem 311(3):1909–1915. doi:10.1007/s10967-016-5155-0
Chaudhuri P, Naskar N, Lahiri S (2017) Measurement of background radioactivity in surface soil of Indian Sundarban. J Radioanal Nucl Chem 311(3):1947–1952. doi:10.1007/s10967-016-5158-x
Jang M, Chung KH, Ji Y-Y, Lim JM, Kim CJ, Kang MJ, Choi GS (2016) Indoor external and internal exposure due to building materials containing NORM in Korea. J Radioanal Nucl Chem 307(3):1661–1666. doi:10.1007/s10967-015-4375-z
Zaim N, Atlas H (2016) Assessment of radioactivity levels and radiation hazards using gamma spectrometry in soil samples of Edirne. Turkey. J Radioanal Nucl Chem 310(3):959–967. doi:10.1007/s10967-016-4908-0
Bourdon B (2016) Uranium Decay Series. In: White WM (ed) Encyclopedia of geochemistry: a comprehensive reference source on the chemistry of the earth. Springer, Cham, pp 1–6
Papadopoulos A, Christofides G, Koroneos A, Stoulos S, Papastefanou C (2013) Radioactive secular equilibrium in 238U and 232Th series in granitoids from Greece. Appl Radiat Isot 75:95–104. doi:10.1016/j.apradiso.2013.02.006
IAEA (2004) Soil sampling for environmental contaminants. IAEA TECDOC No. 1415. International Atomic Energy Agency, Vienna, Austria
Bakshi AK, Prajith R, Chinnaesakki S, Pal R, Sathian D, Dhar A, Selvam TP, Sapra BK, Datta D (2017) Measurements of background radiation levels around Indian station Bharati, during 33rd Indian Scientific Expedition to Antarctica. J Environ Radioact 167:54–61. doi:10.1016/j.jenvrad.2016.11.025
Long SE, Martin TD, Martin ER (1994) Method 200.8 determination of trace elements in waters and wastes by inductively coupled plasma-mass spectrometry, Revision 5.4. U.S. Environmental Protection Agency, Cincinnati, Ohio
UNSCEAR (2000) Sources and effects of ionizing radiation. United Nations Publications, New York
Acknowledgements
The authors would like to thank Jordan Atomic Energy Commission (JAEC) for granting permission to use JAEC laboratory facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Al-Shboul, K.F., Alali, A.E., AL-Khodire, H.Y. et al. Assessment of secular equilibrium and determination of natural and artificial radionuclide concentrations in the zone surrounding the site of the first nuclear reactor in Jordan. J Radioanal Nucl Chem 314, 1353–1360 (2017). https://doi.org/10.1007/s10967-017-5504-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5504-7