
Abstract
The extraction/complexation of uranyl ion by structurally modified sulphoxides in ionic liquid was investigated. These systems were found to be highly efficient, selective and radiolytically stable for hexavalent f-block elements. The extraction proceeds via ‘cation exchange’ mechanism through the species, [U(NO3)·2L]+. Processes were found to be kinetically slow, thermodynamically spontaneous and proceeded via single species. The extended resonance and steric crowding favoured the complexation of phenyl sulphoxides. Na2CO3 was found to be suitable for the quantitative back extraction of uranyl ion from the ionic liquid phase. Finally, these systems were employed for the processing radioactive waste.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to the favourable properties like low vapour pressure, wide liquid range, high degree of solubility, large potential window, high degree of chemical and thermal stability, room temperature ionic liquid finds application in synthesis, electrochemistry, separation, polymer science, catalysis and even in nuclear industries [1–8]. Apart from its ‘green solvent’ image, some interesting chemistry was reported in ionic liquid based systems which otherwise is not conventional [9–11]. The most promising property of ionic liquid is the extraordinary degree of tunability, i.e. the physical and chemical properties of the ionic liquid can be fine tuned by modification of either cationic or anionic part of the ionic liquid. Therefore, sometimes it is called as ‘designer solvent’. In nuclear industry, the main application of ionic liquid was reported as diluent, which in combination with ligand functionality was used for the extraction of actinides and long lived fission products like 90Sr [12–16]. The ligand functionality chemically attached to the ionic liquid, known as task specific ionic liquid was also used for the processing of actinides [17–21]. The kinetics, thermodynamics, extraction mechanism, metal–ligand stoichiometry and the overall complexation chemistry were found to differ in ionic liquid based separation compared to the conventional molecular diluent based separation [10, 14–16]. In recent publications comparative evaluation on the radiolytic stability, extraction efficiency and selectivity revealed the superiority of ionic liquid based systems [13, 22].
On the other hand, the success of any nuclear energy programme largely depends on the effective utilization of the fissile resources both in ‘front end’ and ‘back end’ of the nuclear fuel cycle. Additionally, in back end of the nuclear fuel cycle, the separation is necessary not only in view of recovery of precious fissile isotope but also to avoid the long term surveillance and associated high radiological risk of nuclear waste. Uranium being the key element of nuclear industry, is of high research interest either in separation science or in the basic complexation. Though several functionalities like phosphates, phosphinates, amides, diamides, carbamoyl methyl phosphine oxides, phosphinic acids, diglycolamides etc., in ionic liquid are under investigation for understanding the separation and complexation chemistry of actinides [23–28], yet the sulphoxide moieties in ionic liquid for extraction and complexation of ‘f-block’ elements have not yet been explored.
In view of these above facts, an attempt was made for the efficient, selective and ‘green’ separation process of uranium using structurally modified sulphoxides in room temperature ionic liquid. The study also includes understanding the extraction mechanism, speciation, kinetics, thermodynamics, stripping and radiolytic stability. The complexation of uranyl ion with these sulphoxides in ionic liquid was further investigated by photoluminescence spectroscopy. Finally the above systems were applied for nuclear waste remediation using simulated high level waste (SHLW) solutions originated from research reactor (RR), pressurized heavy water reactor (PHWR) and fast breeder reactor (FBR) [29, 30].
Experimental
Instrument
ICP-AES
The analysis was carried out using Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) with Capacitatively Coupled Device (CCD) as detector system procurred from Spectro Arcos, Germany (Model No: Arcos FHS12 ICP-AES). Operating conditions and instrumental specifications are listed in Supplementary Table 1.
Luminescence
Emission and excitation spectra were recorded on an Edinburgh F-900 Fluorescence Spectrometer in the 200–750 nm region with a Xe lamp as an excitation source, M-300 monochromators and a Peltier cooled photo multiplier tube as detector. The acquisition and analysis of the data were carried out by F-900 software supplied by Edinburgh Analytical Instruments, UK.
Reagents and standard solutions
Standard solutions for all the elements were prepared from CertiPUR® ICP standard reference material solution of individual elements (E-Merck, Darmstadt, Germany) by proper dilution. Supra—pure HNO3 (E-Merck, Darmstadt, Germany) and quartz double distilled water were used throughout the study. Multi-point standardization and five replicate measurements were carried out for all the samples.
Uranyl stock solution was prepared by dissolving spec pure U3O8 in concentrated HNO3. Xylene was procured from Prabhat Chemicals, Gujarat Mumbai, whereas oxalic acid, EDTA and Na2CO3 were produced from Thomas Baker Chemical limited and Qualigens fine Chemicals, Mumbai, India, respectively. Benzyl methyl sulphoxide (BMSO), allyl phenyl sulphoxide (APSO), di isobutyl sulphoxide (DISO) and di hexyl sulphoxide (DHSO) were procured from ICN Pharmaceuticals. Inc, Life Science Group, Plainview.N.Y, Canada. 1-Ethyl-2,3-dimethylimidazolium bis (trifluoromethylsulfonyl)imide and 1-Ethyl-2,3-dimethylimidazolium chloride were procured from Global Nanotech, India. More than 99 % pure ionic liquid solutions were used for further investigation without any purification. Bis (trifluoromethylsulfonyl)imide lithium salt (LiNTf2) has been procured from Aldrich Chemistry, USA. The structures of the ionic liquids and the sulphoxides have been shown in Fig. 1.

Structures of different sulphoxide ligands and ionic liquid diluent
Method
Extraction
For extraction experiment 5 mL of 10 mg L−1 of U solution of required acidity (for feed acidity variation experiments the acidity was varied in the range of 0.01 M HNO3–6 M HNO3 while for other experiments the feed acidity was kept 1 M HNO3) was taken in a glass container. Then 5 mL or organic phase conating modified sulphoxides in ionic liquids were added into it. They were allowed to shake for 2 h in a thermostated water bath at 300 K. After 2 h of shaking, both the phases were allowed to settle for 10 min for complete phase separation. Then suitable aliquots were taken for the fedd into the plasma for ICP-AES analysis. The distribution ratio values for uranyl was expressed by conventional way as follows
The subscripts org and aq refer to the organic and aqueous phases respectively. After extraction of metal ion the raffinate was fed to plasma for the evaluation of D values. For all the ligands, the concentration was optimized as 0.2 M where as the aqueous feed acidity was fixed at 1 M HNO3 (unless it was otherwise mentioned). All the extraction experiments were carried out in a thermostated water bath at 300 K temperature. The initial concentration for uranyl ion taken was 10 mg L−1 for all the extraction experiments.
Stripping
For stripping studies, in the first step the extraction experiments were carried out in optimized experimental conditions to have U-sulphoxide loaded ionic liquid phase. Then the loaded organic phase was separated and allowed to contact with fresh strippant solution.
Variation of imCl and LiNTf2
The experiments carried out using imCl and LiNTf2 are two separate experiments. Basically in one set of experiment, LiNTf2, which is water soluble, is dissolved in various concentration into the aqueous phase containing uranium solution. Then this aqueous phase is allowed to equilibriate with organic phase containing sulphoxide as ligand and imNTf2 as diluent. If the extraction proceeds via anion exchange, mechanism, then the NTf −2 ion from ionic liquid phase would come to aqueous phase in place of thorium extracted species. Due to the presence of NTf −2 ion in aqueous phase coming from LiNTf2, this anion exchange mechanism would expected to be hampered. So the DU value is expected to decrease.
On the other hand the similar experiments were carried out using imCl as aqueous soluble reagent having same cation as that of the water insoluble ionic liquid (imNTf2) used as diluent in the present case. If the cation exchange mechanism predominates, then the im+ cation from organic phase would come to aqueous phase in place of thorium extracted species. Therefore, im+ cation in aqueous phase coming from imCl hampers this mechanism and hence decrease the DU value. Similar approach was also adopted in the Ref. [13].
Radiolytic stability
For radiation stability study, the irradiated organic phase (containg ligands and ionic liquid) was allowed to equlibriate with 10 mg L−1 of U solution in 1 M HNO3 at 300 K for 2 h (with phase ratio = 1:1). Then it was centrifuged for 10 min for complete phase separation. Then the aqueous raffinate was collected and fedinto the plasma for ICP-AES analysis. The D values determined using gamma irradiated solvent systems were compared with that of un-irradiated solvent systems.
Processing of SHLW
For all the experiments (except SHLW), 5 mL of organic phase was allowed to equilibrate with 5 mL of aqueous phase whereas for SHLW, 10 mL of organic phase was allowed to equilibrate with 10 mL of aqueous phase.
Results and discussion
Extraction profile of uranyl
The distribution ratio values for uranyl ion were varied as a function of feed acid concentration for all the sulphoxides in room temperature ionic liquids. The distribution ratio values were found to be an order of magnitude more in case of ionic liquid based systems compared to the molecular diluent based systems [31]. The DU values were found to decrease gradually with increase in feed acidity up to 1 M HNO3. Beyond that a drastic decrease in DU values were obtained for all the cases. This trend was quite different than generally observed for neutral ligands in molecular diluents where the extraction proceeds via ‘solvation mechanism’. The observed trend was found to be similar for ionic liquid based systems reported in the literatures, which suggested that the extraction proceeded via ‘ion exchange’ mechanism [10–13]. The variation of DU as a function of feed HNO3 acid concentration, was found to be still insufficient in understanding whether cation exchange (Eq. 1) or anion exchange predominated (Eq. 2) during uranyl extraction.
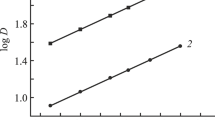
n and m represent the number of sulphoxides molecules and NO3 − ion attached in the extracted species. Im+ and NTf −2 are the constituents cations and anion of the ionic liquid. The trend of the DU values for the above sulphoxides followed the trend APSO > BMSO > DISO > DHSO up to 1 M HNO3. In APSO and BMSO the sulphoxide moieties are in conjugation with the phenyl ring resulting enhanced electron density on the ligating oxygen atom. Moreover, due to the planar structure of the phenyl moiety the approach of the uranyl ion might be easier. In case of hexyl and isobutyl alkyl group substituted sulphoxides the steric factor might be responsible for the weaker complexation and hence decreased DU values. Fig. 2 depicted the extraction profiles of uranyl with structurally modified sulphoxides in room temperature ionic liquid.

The variation of DU values as a function of aqueous feed acidity, ligand conc. 0.2 M, 300 K, Phase ratio 1, time of equilibration 2 h
At higher feed acidity, the H+ ion can also be extracted into the ionic liquid phase by the sulphoxide ligands (Eq. 3). Due to the competition of H+ and uranyl ion, there was a decrease in the distribution of the metal ion with increase in feed acidity.
Extraction kinetics
Time of reaching equilibrium is one of the very important parameters in large scale processing of the metal ion. In the present case the DU values for all the systems were found to increase up to a time of contact of 100 min while beyond that plateau was observed. This suggested that 100 min is required for achieving complete equilibrium. Since all the structurally modified sulphoxides required same time for attaining equilibrium, this kinetics might be an implication of diluent property. The observed kinetics was found to be slower compared to that observed in case of molecular diluent based systems. Similar slower kinetics was also observed in case of uranyl extraction using Bis(2,4,4-trimethyl) pentyl phosphinic acid in C8mimNTf2 compared to that in xylene [13]. The slower kinetics for uranyl extraction in ionic liquid based systems can be attributed to the higher viscosity coefficient of the ionic liquid compared to molecular diluent [14, 19]. The viscous ionic liquid enhances the mass transfer and hence the kinetics. Fig.3 shows the variation of DU values as a function of contact time of both the phases.

The extraction kinetics of uranium at 1 M HNO3 feed acidity,, ligand conc. 0.2 M, 300 K, Phase ratio 1
Determination of Uranyl-sulphoxide stoichiometry and associated thermodynamic constants
To understand the complexation it is required to evaluate the metal–ligand stoichiometry. The equilibrium constants (Kex) for the above Eqs. (1 and 2) can be expressed as [13]
At a particular feed acid concentration, concentration of nitrate ion is constant at constant temperature. Similarly at a particular temperature, the ratio of distribution of either cation or anion in aqueous and ionic liquid phase were also remaining constants. Therefore, the overall equation can be simplified and rearranged in the following fashion written below [13] .
where, \({\text{K}}_{{\text{ex}}}^\prime\) is the conditional extraction constants. Equation 5 implies that a plot of log D versus log[L] should give a straight line with the slope ‘n’, the number of ligand molecules attached to a uranyl ion. The change in Gibb’s energy during the separation of the uranyl ion can be expressed by the following equation [12].
Now if we consider the formation of sulphoxide complexes by uranyl ion in ionic liquid phase then the conditional complex formation constant (kf) can be expressed as [12, 13]
where, pU is the partition function of uranium in aqueous phase and the ionic liquid phase in absence of ligands.
Fig. 4 represents the linear plots of distribution ratio values as a function of ligand concentration. The slope, intercept and the linear regression coefficients were summarized in Supplementary Table 2. This study indicated that two sulphoxide ligands were attached to one uranyl ion. The conditional extraction constants were found to follow the trend APSO > DISO > BMSO > DHSO. The Gibb’s free energy was found to decrease on uranyl extraction suggesting the spontaneity of the separation process. The study also revealed that U-APSO complex was energetically the most favourable followed by U-BMSO, U-DISO and U-DHSO complex. Since the overall energetics of the processes mainly depend on the complexation energy, the complex formation constants also followed the same trend as it was seen for the extraction constants value (Table 1).

The variation of DU values as a function of ligand concentration from 1 M aqueous feed acidity, 300 K, Phase ratio 1, time of equilibration 2 h
Nitrate variation
To ascertain the exact extraction mechanism and nature of species involved in the extraction process, it is required to vary different experimental parameters like, NO3 −, NTf −2 and im+ concentration and their effect on the DU value should be investigated properly. At a particular temperature and particular ligand concentration, Eq (5) can be simplified as
The plots of log D versus logarithm of nitrate ion concentration were shown in Fig. 5. The slope, intercept, associated error, linear regression coefficients were tabulated in the supplementary Table 3. The slope values can provide the number of nitrate anion associated with the metal ion in the extracted species. This study revealed that for all the systems the extracted species was found to be [UO2(NO3)·2L]+ (Fig. 6). This also revealed that ‘cation exchange’ mechanism was predominated during extraction.

The variation of DU values as a function of nitrate ion concentration, 300 K, Phase ratio 1, time of equilibration 2 h

Formation of [UO2(NO3)·2L]+ species and resonance stability of phenyl substituted sulphoxides
Variation of imidazolium ion concentration in aqueous phase
For further confirmation of ‘cation exchange’ mechanism, The DU values were varied as a function of imidazolium cation in the aqueous phase. If Eq (1) is correct then with increase in imidazolium ion concentration in aqueous phase the DU values were expected to decrease. The plots of logD versus log[im+] were found to follow the linear pattern [Supplementary Fig. 1] with slope values ~1. This study revealed that during extraction of uranyl species the overall charge of the extracted complex was 1. Therefore, only one imidazolium ion got exchanged per uranyl species. Supplementary Table 4 summarizes the slope, intercept associated error and the linear regression coefficients of the above plots. Due to very small charge density, the chemical potential for im+ ion will be much smaller than H+ ion and even smaller than UO2 2+ ion. Moreover plenty of im+ ions are there in the organic phase in the form of imNTf2. Therefore, the competition of im+ towards the complexation to sulphoxides compared to uranyl is negligibly insignificant. The [im+] can modify the DU values only if it is cation exchange mechanism i.e. during extraction of uranium species equivalent amount of im+ ion would come to the aqueous phase from the organic phase to maintain the charge neutrality.
Variation of NTf −2 anion in aqueous phase
Again the absence of anion exchange mechanism was reconfirmed by studying the DU values as a function of NTf −2 ion concentration in the aqueous phase. Over a wide range of NTf −2 ion concentration in the aqueous phase no change in the DU value was observed [Supplementary Fig. 2]. This revealed that NTf −2 was not participating in the extracted uranium species and it also reconfirmed the absence of ‘anion exchange’ mechanism.
Time resolved fluorescence spectroscopic investigation of the extracted species
Time resolved fluorescence spectroscopic study is routinely used for probing local environment around metal ion in the extracted complex. Fig. 7 is depicting the emission profiles of different extracted uranyl-sulphoxide complexes in ionic liquid. For U-DISO complex a series of peak was observed at 488.06, 508.70, 526.21 and 548.05 nm with relative intensity 1.43:2.36:1.85:1 which were shfted in case of uranyl complexes of DHSO, APSO and BMSO. The peak positions and their relative intensities were summarized in Supplementary Table 5. The nature of emission profiles for all the three systems were found to be quite different revealing the difference in their speciation. The mono exponential nature of the decay curves for all the systems revealed the presence of single species in the extracted complex whereas the lifetime values for these extracted species was found to be 22.1, 18.9, 33 and 28 µs for DISO, DHSO, APSO and BMSO, respectively. For aqueous nitric acid feed the life time of the uranyl ion was reported to be 15.74 µs [8]. The spacing between these vibronic coupling can be related to the symmetric stretching frequency for uranium oxygen bond in O=U=O moiety [5, 8]. In aqueous system this symmetric stretching frequency was reported to be 822.44 cm−1. On complexation with DISO, DHSO, APSO and BMSO, the symmetric stretching frequency for uranium was found to decrease and became 500, 638, 416 and 429 cm−1, respectively. Due to the approach of the ligands through the equatorial plane the electron density on uranyl uranium decreases and hence the bonds between U and O in uranyl moiety becomes weaker and hence bond length also increases. Similar trend of reduction in stretching frequency on complexation of uranyl ion was reported in case of TOPO complex [8]. For phenyl substituted sulphoxides i.e. BMSO and APSO, the planner structure of the phenyl ring reduces the steric crowding around uranium and hence form stronger complex with sulphoxide oxygens. The stronger bonding between U and sulphoxide oxygen reduces the bond order in uranyl moiety. Therefore, the lowest stretching frequency was observed for APSO and BMSO. In case of alkyl substituted sulphoxides i.e. DHSO and DISO, the steric crowding hampers the approach of the sulphoxide moiety towards uranyl ion and hence lesser perturbation was observed in vibronic stretching frequency values.

The emission profiles for the uranyl complexes of different sulphoxide ligands in room temperature ionic liquid
Radiolytic stability
During processing of radioactive waste the solvent systems are continuously exposed to the high energy particles (α, β) or radiation (γ). Due to the deposition of large amount of energy, the solvent systems may undergo radiolytic cleavage at different parts of ligands or diluents. The nature of cleavage depends on the amount of energy deposition and the nature of the solvent system. This may lead to the degradation in the performance of the solvent systems. An ideal solvent system should be as radio-resistant as possible. In view of this all the present sulphoxide-ionic liquid based solvent systems were exposed to gamma dose of up to 1000 kGy and with the irradiated solvent systems the extraction experiments were carried out to evaluate their radiolytic stability. The overall radiolytic stability for ionic liquid based systems were reported to be more than that of molecular diluents based systems [22, 32]. For DISO and DHSO, the radiation stability was found to be poorer even at 500 kGy exposure. After 1000 kGy of gamma exposure, the DU values became less than 1/5th of their original values in case of alkyl substituted sulphoxides. For phenyl substituted sulphoxides i.e. BMSO and APSO, the radiation stability was found to be very good. Only 15 % of reduction in DU values was observed even after 1000 kGy gamma exposure. The higher radiation stability of the phenyl substituted sulphoxides can be attributed to the resonance stabilization of the phenyl based radical generated on gamma irradiation. Figure 8 represents the changes in DU values as a function of gamma dose for all these four systems.

Radiolytic stability of the sulphoxides in ionic liquid
The main aim of the study was to investigate how the DU values were affected by using gamma irradiated solvent systems. This study is of importance while processing radioactive waste solution. It is expected that on irradiation, there will be breaking of the weakest bonds in the solvent systems and may also lead to radiation induced polymerization. This involves a complex radiation chemistry of ligands as well as diluents. In the context of the manuscript, our aim was to evaluate the solvent systems on the basis of reusability, not on the basic radiation chemistry aspect of these solvent systems which will require a separate investigation. It was also to be noted that there meight be a change in viscosity of the solvent systems on irradiation. The kinetics experiments were carried out for all the solvent systems after gamma irradiation of 1000 kGy. It was observed that with irradiated solvent systems, the kinetics become slightly slower. Within 120 min all the systems were found to rich the equilibrium DU values (Supplementary Figure 3). The DU values specified in Fig. 10 were obtained only after 2 h of equilibration and hence equilibrium DU values.
Stripping of uranium from ionic liquid phase
In view of reusability of the solvent systems, it is essential to strip back the extracted metal ion from the loaded organic phase. Due to appreciable DU values throughout the feed acid concentration, changes in acidity cannot strip uranyl ion from the ionic liquid phase. This necessitates the strippant having aqueous complexation ability with the uranyl ion. A series of such solutions were scanned for such purpose. 0.05 M EDTA, oxalic acid and Na2CO3 were found to strip uranyl ion from the ionic liquid phase effectively (~60 % or more in a single contact). In between these three solutions, oxalic acid was found to be the poorest followed by EDTA. 0.05 M Na2CO3 was found to be the most promising strippant for almost quantitative recovery (more than 99.9 %) of the uranyl ion even in a single contact. Fig. 9 summarizes the stripping behavior of these three strippants for uranyl ion from sulphoxide complexes in ionic liquid. Na2CO3 was also reported to be the most effective strippant for hexavalent actinides from diglycolamides and phosphinic acid complexes in ionic liquid phase [13, 17].

The stripping of uranyl ion from sulphoxide-ionic liquid phase
Selectivity of the solvent systems
The success of any solvent systems lies in the effective application for processing of actual waste solutions. In this context, high level waste solutions of research reactor, FBR and PHWR grade high level synthetic waste solutions [29, 30] were processed with these sulphoxides based ligands in ionic liquid. Table 2 summarizes the results for the best solvent systems i.e. APSO in ionic liquid while that for BMSO, DHSO and DISO are summarized in Supplementary Table 6, 7, and 8, respectively. The analytical results revealed that these solvent systems are highly selective for hexa-valent actinide ions while other metal ions (Sr, Y, Rb, Nb, Pd, Ag, Cd, Ba, Pr, La, Nb, Sm, Eu, Gd, Dy, Cr, Fe, Mn, Ni, Na, Ca and Al) are almost un-extracted by these solvent systems. Supplementary Table 9 summarizes the initial and final concentrations of uranium while processing the SHLWs using different structurally modified sulphoxides in ionic liquid.
Conclusions
Different structurally modified sulphoxides in room temperature ion liquid were found to be highly efficient and selective solvent systems for hexa and tetra valent actinide ions with a trend of APSO > BMSO > DISO > DHSO. The resonance of phenyl ring enhances the electron density on sulphoxide O enhancing the ligating ability whereas the alkyl substituents on sulphoxides enhance the steric crowding reducing the extraction efficiency of uranyl ion. The overall extraction process was found to be thermodynamically favorable proceeding through ‘cation exchange mechanism with [UO2(NO3)·2L]+ species. The extraction process was found to be kinetically slower attributed to the viscosity effect of the ionic liquid. 0.05 M Na2CO3 was found to be very effective for almost quantitative recovery of uranyl ion. The phenyl substituted sulphoxides i.e. APSO and BMSO in ionic liquid were found to be highly radio-resistant. Finally, APSO in ionic liquid was found to be very effective in processing SHLW solutions of research reactor, PHWR and FBR origins.
References
Peia Y, Wanga J, Wua K, Xuan X, Lu X (2009) Ionic liquid-based aqueous two-phase extraction of selected proteins. Sep Purif Technol 64(3):288–295
Soto A, Arcea A, Khoshkbarchi MK (2005) Partitioning of antibiotics in a two-liquid phase system formed by water and a room temperature ionic liquid. Sep Purif Technol 44(3):242–246
Park S, Kazlauskas RJ (2003) Biocatalysis in ionic liquids—advantages beyond green technology. Current Opinion in Biotechnol. 14(4):432–437
Sengupta A, Murali MS, Mohapatra PK, Iqbal M, Huskens J, Verboom W (2015) An insight into the complexation of UO22 + with diglycolamide-functionalized task specific ionic liquid: kinetic, cyclic voltammetric, extraction and spectroscopic investigations. Polyhedron 102:549–555
Sengupta A, Murali MS, Mohapatra PK (2013) Role of alkyl substituent in room temperature ionic liquid on the electrochemical behavior of uranium ion and its local environment. J Radioanal Nucl Chem 298:209–217
Sengupta A, Murali MS, Mohapatra PK, Iqbal M, Huskens J, Verboom W (2014) Extracted species of Np(IV) complex with diglycolamide functionalized task specific ionic liquid: diffusion, kinetics and thermodynamics by cyclic voltammetry. J Radioanal Nucl Chem. doi:10.1007/s10967-014-3857-8
Sengupta A, Murali MS, Mohapatra PK (2014) Electrochemical behavior of Cerium (IV) in a RTIL and its mixture with ethanol. J Rare Earths 32(7):641–647
Mohapatra PK, Raut DR, Sengupta A (2014) Extraction of uranyl ion from nitric acid medium using solvent containing TOPO and its mixture with D2EHPA in room temperature ionic liquids. Sep Purif Technol 133:69–75
Sengupta A, Mohapatra PK, Iqbal M, Huskens J, Verboom W (2012) A highly efficient solvent system containing functionalized diglycolamides and an ionic liquid for americium recovery from radioactive wastes. Dalton Trans 41(23):6970–6979
Rout A, Venkatesan KA, Srinivasan TG, Vasudeva Rao PR (2010) Unusual extraction of plutonium(IV) from uranium(VI) and americium(III) using phosphonate based task specific ionic liquid. Radiochim Acta 98(8):459–466
Giridhar P, Venkatesan KA, Subramaniam S, Rao PRV (2008) Extraction of uranium (VI) by 1.1 M tri-n-butylphosphate/ionic liquid and the feasibility of recovery by direct electrodeposition from organic phase. J Alloys and Comp 448(1–2):104–108
Mohapatra PK, Sengupta A, Iqbal M, Huskens J, Verboom W (2013) Diglycolamide-functionalized calix[4]arenes showing unusual complexation of actinide ions in room temperature ionic liquids: role of ligand structure, radiolytic stability, emission spectroscopy, and thermodynamic studies. Inorg Chem 52(5):2533–2541
Singh M, Sengupta A, Murali MS, Kadam RM (2016) Selective separation of Uranium from nuclear waste solution by Bis(2,4,4-trimethyl) pentyl phosphinic acid in ionic liquid and molecular diluents: a comparative study. J Radioanal Nucl Chem. doi:10.1007/s10967-016-4691-y
Sengupta A, Mohapatra PK (2012) Extraction of radiostrontium from nuclear waste solution using crown ethers in room temperature ionic liquids. Supramol Chem 24(11):771–778
Visser AE, Rogers RD (2003) Room-temperature ionic liquids: new solvents for f-element separations and associated solution chemistry. J Solid State Chem 171(1–2):109–113
Lertlapwasin R, Bhawawet N, Imyim A, Fuangswasdi S (2010) Ionic liquid extraction of heavy metal ions by 2-aminothiophenol in 1-butyl-3-methylimidazolium hexafluorophosphate and their association constants. Sep Purif Technol 72(1):70–76
Sengupta A, Mohapatra PK, Iqbal M, Huskens J, Verboom W (2013) A diglycolamide-functionalized task specific ionic liquid (TSIL) for actinide extraction: solvent extraction, thermodynamics and radiolytic stability studies. Sep Purif Technol 118:264–270
Paramanik M, Raut DR, Sengupta A, Ghosh SK, Mohapatra PK (2016) A trialkyl phosphine oxide functionalized task specific ionic liquid for actinide ion complexation: extraction and spectroscopic studies. RSC Adv. 6:19763–19767
Mohapatra PK, Sengupta A, Iqbal M, Huskens J, Verboom W (2013) Highly efficient diglycolamide-based task specific ionic liquids: synthesis, unusual extraction behaviour, irradiation, and fluorescence studies. Chem A Euro J 19(9):3230–3238
Ouadi A, Klimchuk O, Gaillard C, Billard I (2007) Solvent extraction of U(VI) by task specific ionic liquids bearing phosphoryl groups. Green Chem 9:1160–1162
Biswas S, Rupawate VH, Roy SB, Sahu M (2014) Task-specific ionic liquid tetraalkylammonium hydrogen phthalate as an extractant for U(VI) extraction from aqueous media. J Radioanal Nucl Chem 300(2):853–858
Singh M, Sengupta A, Murali MS, Kadam RM (2015) Comparative study on the radiolytic stability of TBP, DHOA, Cyanex 923 and Cyanex 272 in ionic liquid and molecular diluent for the extraction of thorium. J Radioanal Nucl Chem. doi:10.1007/s10967-015-4624-1
Mudring AV, Tang S (2010) Ionic liquids for lanthanide and actinide chemistry. Euro. J Inorg Chem. 18:2569–2581
Sun X, Luo H, Dai S (2013) Ionic liquids-based extraction: a promising strategy for the advanced nuclear fuel cycle. Chem Rev 112:2100–2128
Vasudeva Rao RR, Venkatesan KA, Rout A, Srinivasan TG, Nagarajan K (2012) Potential applications of room temperature ionic liquids for fission products and actinide. Sep Sci Technol 47:204–222
Dietz ML (2006) Ionic liquids as extraction solvents: where do we stand? Sep Sci Technol 41:2047–2063
Cocalia VA, Jensen MP, Holbrey JD, Spear SK, Stepinski DC, Rogers RD (2005) Identical extraction behavior and coordination of trivalent or hexavalent f-element cations using ionic liquid and molecular solvents. Dalton Trans 11:1966–1971
Rout A, Venkatesan KA, Srinivasan TG, Vasudeva Rao PR (2012) Liquid–liquid extraction of Pu(IV), U(VI) and Am(III) using malonamide in room temperature ionic liquid as diluents. J Hazard Mater 221–222:62–67
Sengupta A, Murali MS, Thulasidas SK, Mohapatra PK (2014) A novel solvent system containing CMPO as the extractant in a diluent mixture containing n-dodecane and isodecanol for actinide partitioning runs. Hydrometallurgy 147–148:228–233
Prabhu DR, Sengupta A, Murali MS, Pathak PN (2015) Role of diluents in the comparative extraction of Th(IV), U(VI) and other relevant metal ions by DHOA and TBP from nitric acid media and simulated wastes: reprocessing of U-Th based fuel in perspective. Hydrometallurgy 158:132–138
Shukla JP, Kedari CS (1996) Influence of the nature of organic diluents on the extraction of uranium(vi) by bis(2-ethylhexyl) sulfoxide from nitric acid solutions. J Radioanal Nucl Chem 207(1):93–105
Sengupta A, Mohapatra PK, Patil AB, Kadam RM, Verboom W (2016) Radiation stability of diglycolamide functionalized calix[4]arenes in ionic liquid: solvent extraction, EPR and GC–MS studies. Sep Purif Technol 162:77–83
Acknowledgments
The author wish to acknowledge Dr. R.M.Kadam, Head, Actinide Spectroscopy Section and Dr. P.K.Pujari, Head, Radiochemistry Division for their constant support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Priya, S., Sengupta, A. & Jayabun, S. Understanding the extraction/complexation of uranium using structurally modified sulphoxides in room temperature ionic liquid: speciation, kinetics, radiolytic stability, stripping and luminescence investigation. J Radioanal Nucl Chem 310, 1049–1059 (2016). https://doi.org/10.1007/s10967-016-4970-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4970-7