
Abstract
Age-dating of 137Cs ceramic sources is shown to be a viable technology for nuclear forensics investigations. The 137Cs age-dating method in general, however, could be substantially improved by using radiometric rather than ICP-MS measurement of the 137Cs isotope and by refining the Cs/Ba separation process. With these improvements, uncertainty in the age of a 60-year-old source decreases from 1.35 to 0.68 y.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The date on which a radioactive source material was last separated from its decay daughters is a useful parameter in deducing the material’s origins and history for nuclear forensics investigations. In general, the date of interest is obtained by measuring the relative amounts of the parent radionuclide and its daughter isotopes in a sample of the source material on the date when the sample is analyzed and using the daughter/parent ratio to calculate the time that would have elapsed since the daughter was absent according to the appropriate decay formula. The elapsed time is commonly called the “age” of the material and the process of determining its value is known as “age dating.”
Cesium-137 sealed sources are some of the most popular and widely used radioactive sources for medicine and industry because of the favorable energy of their gamma emission and advantages associated with the nuclide’s 30-year half-life. Because these sources are so broadly available, concerns have long existed that a 137Cs source might be incorporated into a radioactive dispersal device (RDD) for malevolent purposes. In recognition of this situation, the nuclear forensics community has investigated methods for age dating 137Cs materials for a number of years. Early methods [1, 2] were developed for sources containing encapsulated 137CsCl salt because this configuration was especially common in older radiation devices. Expanded understanding of the consequences of a 137CsCl RDD prompted restrictions on the use of that material and a global shift among manufacturers of 137Cs sources to other chemical forms of 137Cs [3]. Sources in which the 137Cs is incorporated into glass or ceramic matrices are common. Logically, accurate age-dating methods for 137Cs sources containing these alternative materials are needed. In prior work [4], we developed a method for Cs-loaded glass. In the present work, age dating of a 137Cs ceramic was investigated.
The process by which the age of a source material is typically determined involves three steps. First, a sample of the material is brought into solution to provide a representative, uniform, and easily dividable sample configuration amenable to subsequent chemical manipulations and measurements. Next, if necessary, measured portions of the source solution are treated to isolate individual constituents of the solution to facilitate measurement of the parent and daughter nuclides. Finally, the parent and daughter nuclides are measured in a way that allows accurate determination of their amount ratios for input to the model decay formula and calculation of the time since purification, as the age of the material is also called.
With 137Cs, the parent radionuclide decays by beta-particle emission to metastable 137mBa, which in turn decays very quickly (half-life of 2.552 min) to stable 137Ba. Assuming no 137Ba is present at t = 0 (consistent with the condition that defines the purification time), the conversion from 137Cs to 137Ba follows the relationship
where \( N_{{^{137} {\text{Ba}}}} \left( t \right) \) is the atom amount of 137Ba in a selected portion of the source material at time t, \( N_{{^{137} {\text{Cs}}}}^{0} \) is the atom amount of 137Cs present at zero time in the same portion of material, λ is the 137Cs decay constant (given by ln 2 divided by the half-life, or 0.02305 y−1), and t is the time elapsed since \( N_{{^{137} {\text{Ba}}}} \) had the value zero. This equation can be solved for the time of decay (i.e., the “age”) and written in terms of the atom amounts of each nuclide present at time t if one recognizes that the sum of the 137Ba atoms generated during the decay and the 137Cs atoms that remain is constant and equal to the atoms of 137Cs at t = 0 (i.e., \( N_{{^{137} {\text{Cs}}}}^{0} = N_{{^{137} {\text{Ba}}}} \left( t \right) + N_{{^{137} {\text{Cs}}}} \left( t \right) \)). The solution provides the basis for calculating the assigned age from measured values and is given by
Age dating a 137Cs source thus requires accurate determination of the 137Ba daughter and the residual 137Cs parent in a sample of the source material so a time of decay can be calculated. The 137Cs in a sample can be measured by either a radiometric technique (gamma spectrometry) or using mass spectrometry. The stable 137Ba isotope requires mass spectrometric measurement. Because the nuclides that must be measured (137Cs and 137Ba) are isobaric, chemical separation of cesium from barium is required for mass spectrometric determination of the isotope amounts. In prior work [1, 2, 4], this separation was made using an extraction chromatography resin (Sr Resin from Eichrom Technologies) and the gas-pressurized-extraction-chromatography (GPEC) apparatus developed by Idaho National Laboratory [1]. After separation, the isotopes in solution were determined by inductively coupled plasma mass spectrometry (ICP-MS).
One difficulty we encountered in pursuing investigation of an age-dating method for the 137Cs ceramic matrix was determining a representative composition for designation as the “ceramic.” We discuss the range of materials that might have been used in fabricating 137Cs ceramic sources and describe considerations that led us to select a cesium-loaded aluminosilicate as the ceramic composition most likely to be encountered in an interdicted 137Cs source or following a 137Cs RDD event. Then, we describe our preparation of a surrogate aluminosilicate ceramic for use in evaluating an age-dating method.
Subsequently, we address the different component steps in applying the age-dating method, including dissolution of the ceramic, separation of its cesium and barium constituents, and ICP-MS analysis of the separated fractions.
Evidence is presented that shows our dissolution procedure to be effective both in decomposing the sample matrix and allowing for quantitative recovery of the cesium and barium constituents. The GPEC method for separating cesium and barium in the dissolved matrix is shown to exhibit some inadequacies, not only in this application but possibly in previous applications as well. An alternative separation approach using a more conventional column configuration but with pumped eluents is proposed and evaluated. Refinement of the separation conditions for both the GPEC and conventional column approaches to use an alternative eluent for barium recovery is recommended. Strategic operational design for sample preparation and our recently developed [5] data-reduction model for correcting non-linearity and mass bias were applied in producing the ICP-MS data on which our evaluations and conclusions are based. The precision and accuracy afforded by this approach to the measurements allowed high confidence in the conclusions reached.
Finally, we address uncertainties associated with the various operations and measurements in the age-dating process and combine the various uncertainty contributions [6] to project the uncertainty associated with the measured age of a 137Cs source material.
Experimental
Considerations in approach to synthesis of surrogate ceramic
To produce their radioactive sources, sealed-source manufacturers have been purported to incorporate 137Cs into various types of ceramic matrices. Some commonly mentioned ceramics for immobilizing 137Cs are zeolites soaked with 137CsCl or ceramics glazed with a specific chemical form of 137Cs (nitrate, sulfate, oxide) [7–10], sodium aluminosilicate mixed with 137CsCl and sintered and/or ion exchanged to create a pollucite phase [11, 12], and sodium-zirconium-phosphate-based ceramics containing 137CsCl [13, 14]. Despite common conjectures made in the literature regarding materials that might be used by sealed-source manufacturers for 137Cs source materials, we found actual evidence in the open-literature only for aluminosilicate-types of cesium ceramic having been used. Thus, we concluded that an aluminosilicate ceramic is the most likely material to be encountered in the event that a 137Cs ceramic source should be interdicted. Consequently, it was decided that a cesium-containing aluminosilicate would be an attractive material on which to base the targeted age-dating method.
Although selection of the aluminosilicate ceramic for the ceramic simulant was a clear choice, choosing the method by which the cesium aluminosilicate ceramic would be fabricated was not as straightforward. Procedures to create generic cesium-containing aluminosilicates do exist in the open literature with sufficient detail for replication. However, published procedures specific to the fabrication of commercial 137Cs ceramic sealed sources are virtually nonexistent, most likely because such procedures are regarded as proprietary information or trade secrets and are rarely released publicly by the manufacturer. After considering alternative options, we chose to fabricate a ceramic surrogate via sodium aluminosilicate, loading the material by ion-exchange with known amounts of Cs and Ba from certified spectroscopic standards. We also elected to use the loaded powder form of the ion-exchanged base material as our ceramic surrogate. The reasoning behind this choice of fabrication approach was two-fold. First, our chemists, based on considerable experience with aluminosilicate materials, predicted that there should be little difference in the dissolution behavior between the ion-exchanged aluminosilicate precursor and any ceramic form produced from it by sintering, as long as hydrofluoric acid were used to decompose the silicate matrix. Traditional ceramics are generally fired at a temperature that transforms the material from powder to a ceramic monolith [15]. With some types of ceramics, the chemical properties of the starting material are quite different from the fired product. For example, γ-alumina powder dissolves in strong acids and bases while sintering the powder to a ceramic produces α-alumina, which is very much more resistant to chemical attack. With cesium aluminosilicates, a fired ceramic is expected to dissolve almost as readily as the precursor powder in an acid mixture containing HF, HNO3, and HCl. Thus, firing the precursor powder to a ceramic would not be required for the purpose of developing a dissolution method.
The second factor we considered was an ability to control the chemical composition of the product. When developing a dissolution and separation procedure for age dating a cesium ceramic source, a particularly useful measure of the quality of the developed processes is quantitative recovery of the cesium and barium originally present in the material. To reliably evaluate the recovery, it is desirable to know the amounts of cesium and barium present in each sample with a high level of certainty. Numerous reports in the literature for cesium ceramics studied as chemical waste forms indicate volatilization losses of cesium in the ceramic at certain firing temperatures, up to as much as 4 % of the initial cesium loading [9, 10]. By firing our surrogate, we would introduce an unnecessary source of uncertainty in the amount of cesium present in the ceramic, and make evaluation of recovery more difficult. Along these lines, we did investigate the possibility of using a cesium-containing aluminosilicate that was previously manufactured in our facilities for evaluation as a potential waste form matrix [10]. We concluded that the levels of impurities and extraneous elements in the material, as well as uncertain amounts of cesium and barium, would produce unnecessary complications for the dissolution procedure (e.g., incomplete dissolution of elements in the waste form that likely would not even be present in an actual 137Cs ceramic sealed source) and limit the accuracy of recovery estimates. The fabrication approach based on soaking a sodium aluminosilicate with cesium and barium and creating the ceramic simulant via ion exchange at room temperature mitigates the risk of volatilizing cesium due to firing the ceramic, produces a surrogate that is anticipated to have representative dissolution properties, and at the same time allows exacting control over the chemical composition of samples produced for study. Our procedure for fabricating the surrogate is provided in the next section.
Procedure for Surrogate Preparation
A high-purity (99.95 % metals basis, Pfaltz and Bauer) sodium aluminum silicate was chosen as the base material for fabrication of the ceramic surrogate because the sodium cation is easily replaced with cesium or barium by ion exchange and the material is a likely starting material in a manufacturing process due to its low cost and abundant availability. Individual portions (nominally 0.1 g) of the sodium aluminum silicate were weighed into 30-mL, pre-cleaned, PTFE beakers. Next, equal weighed portions of cesium and barium spectroscopic standard solutions (High Purity Standards, Charleston, SC; Inorganic Ventures, Christiansburg, VA; or SCP Science, Champlain, NY) were dispensed into each beaker. The mixtures were gently swirled and placed under a laminar-flow hood to air dry overnight. This process allowed ample time for the cesium and barium ions to exchange with the sodium ions present in the aluminosilicate and create the cesium-containing ceramic surrogate. With this preparation, each portion of surrogate is expected to contain accurately known amounts of cesium and barium determined by the mass of standard solution that was added. A sodium aluminum silicate blank was prepared (by adding deionized water in place of the element solutions) and similarly air-dried overnight to allow assessment of barium contamination that might have been introduced during handling of the samples or from the reagents used in the synthesis. After drying, the beakers containing the synthesized ceramic-surrogate solids were covered with Parafilm™ and placed in storage for subsequent use as test samples for evaluating the dissolution and separation procedures.
Dissolution procedure
Given the similarity in chemical composition between the silica-based glass material that was studied in our past work [4] and the aluminosilicate ceramic surrogate, we applied a comparable dissolution method. Silicates in the sample are decomposed with HF and silicon is evolved as SiF4 gas in the first step of the dissolution, which involved adding 2 mL of reagent water, 2 mL of concentrated nitric acid (Optima™ grade, Fisher), 1 mL of concentrated hydrochloric acid (Optima™ grade, Fisher), and 1 mL of concentrated hydrofluoric acid (Optima™ grade, Fisher), covering the beaker with a Teflon® watch glass, refluxing the solution on a hotplate at medium heat for 0.5 h, and then taking each sample to dryness. This step is repeated to ensure that all silicates are decomposed and all silicon is expelled from the sample. Residual fluoride is evolved as BF3 gas by adding 2 mL of a 2 % solution of H3BO3 (Puratronic, 99.9995 % metals basis, Alfa Aesar®) and an additional 2 mL of concentrated nitric acid, taking to dryness, and repeating to ensure all excess fluoride is removed. Excess boric acid is evolved as gaseous trimethyl borate to limit the amount of total dissolved solids introduced into the ICP-MS plasma when samples are ultimately analyzed. To remove the boric acid, 5 mL of Optima™-grade methanol (Fisher Scientific) is added; then, the beaker is covered with a Teflon® watch glass and the liquid is refluxed on a hot plate at 70 °C for a short time (15 min), at which time the watch glass is removed and the solvent allowed to evaporate to dryness. This step is then repeated once more to ensure complete removal of the boric acid. We note that previous development work on this procedure showed that heating the methanol/boric acid mixture at too high a temperature (>80 °C) or in the presence of water can cause disintegration of the trimethyl borate and impede complete boron removal [4, 16]. Salts in each beaker are finally dissolved in 3 M nitric acid, with no visible residues of undissolved solids.
Separations using Sr Resin in an Omnifit® Column Assembly
For our separation tests with the Omnifit® column assembly, Benchmark kits with 3-mm i.d. × 50- and 100-mm long “microbore” columns were procured. The size of the column was selected using capacity specifications for the Sr Resin (Eichrom Technologies website) and a projected loading of 1 µg Cs and 1 μg Ba for mixtures to be tested. The Omnifit® column consists of a glass column tube with fritted, screw-on fittings at each end. Both end fittings connect to narrow bore polyethylene tubing used for bringing solution to and away from the column. The column was mounted in a clamp on a ring stand and the tube from the bottom of the column was connected to a small peristaltic pump with a reverse switch that allowed moving liquid through the column in either the top-to-bottom or bottom-to-top direction. The setup is shown in Fig. 1.

Omnifit® column assembly with 3-mm × 100-mm column
To pack the column, Sr resin (50–100 µm particle size) was slurried in reagent water and the slurry added to the top end of the glass column with the top fitting removed. The resin was allowed to settle and supernatant liquid was pumped out the bottom of the column. Then, more slurry was added and the process was repeated until the entire length of the column was packed. The top fritted fitting was then attached, completing the column preparation. The bed volume was calculated as approximately 0.35 mL for the 50-mm column.
Prior to use, the column was washed by pumping through it (bottom to top) 10 mL of reagent water, 10 mL of 3 N HNO3, and 10 mL of 1 % acetic acid. Then the column was conditioned with another 5 mL of 3 N HNO3. When the column was not in use, it was flushed with DI water and left filled with water. It was reconditioned with 5 mL of 3 N HNO3 just prior to using it for each subsequent separation. Except for packing the column, where solutions were pumped top-to-bottom, all operations were done with the pump set for bottom-to-top liquid flow.
We devised a means to add the sample solution as a small plug of liquid to the system. Starting with a 2 µg/g sample solution (3 N HNO3) in a centrifuge cone, we pumped 0.5 mL of the solution from the tube, stopped the pump, moved the input tube to a tube containing 3 N HNO3, and continued the elution. We determined the exact amount of sample solution transferred to the column by weighing the sample cone before and after the sample was removed. After the sample was loaded in a typical test, we pumped 5 mL of 3 N HNO3 through the column and then put the input tube in a container of 1 % acetic acid and pumped 5 mL of that solution through. Pumping was regulated at 0.5 mL/min. We collected 0.5- or 1.0-mL fractions of the column effluent in centrifuge cones that contained previously measured portions of Pr internal standard. We weighed each tube to determine the exact amount of eluent collected in each fraction and then diluted the fractions for ICP-MS determination of their Cs and Ba contents. Results of these measurements were plotted as elution profiles for Cs and Ba as illustrated in Fig. 2.

Profiles of cesium and barium elution from a 3-mm id × 50-mm long Omnifit® Microbore Column packed with Sr resin: 0.5 mL sample in 3 N HNO3; washed with 5 mL 3 N HNO3 and then 5 mL 1 % HOAc
Inductively coupled plasma mass spectrometry
Instrument
ICP-MS isotope ratio and elemental concentration measurements were made with a Perkin-Elmer/SCIEX™ ELAN DRC-II® quadrupole ICP-MS (Perkin Elmer, Norwalk, CT) equipped with an SC-2-DX auto-sampling system (ESI, Inc., Omaha, NE) and a discrete dynode detector. An all Teflon® sample-introduction system, consisting of a Peltier-cooled PFA-Teflon® cyclonic spray chamber (Model PC3, ESI, Inc.) and low-flow self-aspirating PFA-Teflon® nebulizer (ESI, Inc.) were used to introduce samples into the plasma. We found that the Peltier-cooled spray chamber substantially improved the precision of replicate measurements during analysis of a given sample and strongly recommend its use for isotope ratio measurements. All samples were run in pulse-counting mode with a dead-time-correction setting in the Perkin Elmer software equal to 62 ns. All data sets were run in quantitative-analysis mode. The instrument analysis method was set up with defined parameters of 100-ms dwell time, 15 sweeps per replicate, and 7 replicates per sample for all analysis runs.
All reagents used for ICP-MS dilutions were prepared in FEP-Teflon® bottles due to their superior leach-resistance properties. Certified-sterile polypropylene centrifuge vials (Corning® Centristar™) were used exclusively for sample containment due to their consistently negligible elemental background levels.
Data handling for ICP-MS measurements
The ICP-MS instrument output in units of counts per second for each measured peak was exported to a Microsoft® Excel® workbook so it could be readily manipulated to refine the dead-time correction being applied, to implement corrections for background or interference signals, and to calculate mass-bias corrections from ratios obtained with isotopic standards interspersed among the samples in a given analysis sequence. The first operation applied in treating the data was to remove the dead-time correction that the instrument had applied to the output data so the data set included only detector count rates with no dead-time corrections applied. Then, the data were corrected for background contributions to individual peaks, primarily those from Xe isotopes, and the corrected peak-intensity data for barium standards analyzed during the run were fit to a model that provided a best-fit evaluation of linearity- and mass-bias-correction parameters for the ICP-MS run. The model that was applied to evaluate and correct for the non-linearity and mass-bias effects on the measured isotopic signals is described elsewhere [5].
Measuring cesium and barium isotopes by ICP-MS
Samples for the ICP-MS determination of cesium and barium were prepared within the context of a purposeful analysis strategy intended to reduce random and systematic error contributions to the analysis results by closely controlling additions of internal standard, using ratio-based data reduction and calibration formulas, and employing mass-based measurements throughout, as recommended by Salit and coworkers at NIST [17–19]. Calibration standards for concentration measurements were prepared by mixing weighed amounts of Cs and Ba spectroscopic standard solutions (1000 µg/g diluted by mass to nominally 2 µg/g) with a weighed amount of a nominally 1-µg/g solution of praseodymium, designated as internal standard. The amounts of Cs and Ba standards were selected to produce concentrations of 0, 10, 20, and 30 ng/g when the Pr was diluted to 10 ng/g. In preparing an unknown sample for comparison to these standards, a portion of the sample was transferred to or collected in a weighed tube that contained a weighed portion of the same Pr solution that was used in making the standards, and the mass of the added sample was measured. Samples were then diluted by mass to produce a concentration close to 10 ng/g for the Pr internal standard. By following this protocol, one does not have to know the concentration of the Pr internal standard because the amount of Pr is proportional to the known mass of internal standard solution and, if calibrations and calculations are based on the internal standard solution mass, the concentration cancels between standards and samples. Operations in any given test were laid out ahead of time to specify amounts of internal standard, sample, and diluent to give final concentrations within a narrow range of values comparable to the standards. Most often, the sample tubes were set up to contain 0.5 g of the Pr solution and were brought to a nominal value of 50 g solution after being diluted for analysis. The mass of Pr solution, mass of added sample, and final dilution mass were recorded for every sample. We also monitored the mass of the Pr internal standard stock solution to ensure that its concentration was not changing as a result of evaporation during storage as this would spoil the comparability between samples and standards on which the analysis strategy depends. We did not see any evidence of evaporation over the period of interest but mention that corrections can be applied for small changes in the internal standard solution if evaporation were to be noted.
Each analysis run was set up in a sequence that included the cesium and barium standard solutions. Barium isotopes in the standards were used for evaluating non-linearity and mass-bias effects. The run sequence typically prescribed running the standards, a set of analytical samples, the standards, more samples, and so on, such that every group of analytical samples was bracketed by the standards. The entire standard sequence was usually repeated at least three times during a run so at least three replicate measurements of each desired signal ratio were produced. Blank samples consisting of the 2 % HNO3 diluent were analyzed before and after every sample and standard to ensure complete washout of the sample and prevent carryover of analyte from one sample to the next.
Peaks located at m/z 129(Xe), 130(Xe,Ba), 131(Xe), 132(Xe,Ba), 133(Cs), 134(Xe,Ba), 135(Ba), 136(Xe,Ba), 137(Ba), 138(Ba), and 141(Pr) were measured. The signal at m/z 129 (129Xe) was used to monitor the level of xenon impurity in the argon plasma gas and to allow correction for contributions by xenon isotopes to the signals for 130Ba, 132Ba, 134Ba, and 136Ba. Corrections for the xenon isotopes were made using the ratios to 129Xe observed in the blanks that bracketed each sample and the 129Xe observed for a specific sample because both the level of xenon impurity and its isotopic composition might have varied both within each run and between runs. This variation was a concern because xenon concentrates in the argon supply tank as the argon is used and the xenon isotopes evaporate at slightly different rates, which could cause the xenon isotopic composition to change due to isotope fractionation as evaporation occurs.
Non-linearity (τ) and mass-bias (F) correction factors as defined in Ref. 5 were determined separately for every run using results from the barium standards analyzed during the run and reference ratios for 130/138, 134/138, 136/138, and 137/138 based on the IUPAC abundances for the barium isotopes [20]. We found that, due to the very low abundance of 132Ba (0.1012 %) and the high abundance of 132Xe (26.89 %), corrected 132Ba/138Ba ratios were not very precise; consequently they were not used in modeling the data to determine corrections. In every ICP-MS run that was carried out as part of this task, the fit of data to the model was excellent with corrected ratios of the Ba isotopes to 138Ba deviating not more than 0.2 % from the reference values.
Calibration of the ICP-MS signal response vs concentrations of standards was established from the simple straight-line relationship between the isotope/internal standard signal ratio and the amount ratio of the isotope to internal standard solution in the calibrants. The standard uncertainties of the ICP-MS concentration measurements of Cs (based on 133Cs) and Ba (based on 138Ba) were estimated directly by pooling standard deviations from analysis runs carried out on seven different days during the study. It is convenient to describe the uncertainty in the measurements as arising from two sources: (1) a variance due to background fluctuations and other sources of signal noise, which dominates when the signal is very small and (2) a variance that is proportional to the magnitude of the signal, which becomes most important when the signal is much larger than the noise. Representing the variance from the noise as u 2Noise and the proportional variance as \( C_{\text{Ba}}^{2} u_{\text{Prop}}^{2} \), one can express the variance, \( u_{\text{Ba}}^{2} \), for a barium isotope signal of any magnitude as
Values for u Noise and u Prop for Cs and Ba are shown in Table 1. The proportional uncertainties include contributions from not only sample-to-sample variations, but also from long term drift in the calibration slopes over the several hour duration of the ICP-MS runs. In the case of Cs, this long term drift is a substantial component of the variance while for Ba it is much less significant.
Results and discussion
Quantitative evaluation of dissolution performance
Quantitative performance of the dissolution procedure was tested by analyzing solutions prepared by dissolving a set of surrogate ceramic samples and comparing results of the analysis with values expected from data recorded when the surrogate samples were prepared. Four surrogate samples were made using a nominal mass of 100 mg of sodium aluminosilicate and spectroscopic standard solutions. Components of each surrogate are listed in Table 2.
Each of the four surrogate samples was dissolved using our listed procedure and the solution was transferred from the dissolution beaker to a weighed 50-mL centrifuge vial (Corning™ certified sterile polypropylene vial) using 3 N HNO3. The solution in each vial was brought to a mass of approximately 50 g using additional 3 N HNO3, and the vials were weighed again to determine the total solution mass. The stock solutions prepared in this way were serially diluted by mass to provide a final concentration on the order of 2 µg of Cs or Ba per g of solution for samples to which Cs and Ba had been added; the same dilutions were applied to the unspiked sample, Ceramic 4. Next, 0.5 g of each 2 µg/g solution was weighed into a pre-weighed 50-mL centrifuge cone containing a weighed portion (0.5 g) of a 1 µg/g solution of Pr internal standard. The Pr solution was from the same batch that was used in preparing Cs and Ba calibration standards for ICP-MS determination of the elements. These operations produced analysis solutions containing roughly 20 ng/g concentrations of Cs and Ba, and 10 ng/g Pr.
The solutions spiked with internal standard and the corresponding calibration standards were analyzed in four ICP-MS runs carried out on separate days. Each run provided a value for the concentration of Cs and the concentration of Ba in the solution. Results of these measurements are shown in Table 3.
The precision of the concentration measurements was quite good, having a pooled rsd of 0.26 % for Cs and 0.29 % for Ba. This level of precision for measurements with the quadrupole ICP-MS reflects the effectiveness of the experimental design and data-reduction process incorporated into our operations.
From these measured concentrations and dilution factors calculated from the solution mass data recorded during the dissolution and serial dilutions, we calculated values for the amounts of Cs and Ba recovered from the dissolution process and compared these to the amounts that were added when the surrogate ceramics were prepared (Table 4).
The data show that 98 % or more of the cesium in each dissolved sample was transferred out of the beaker for measurement. Also, the barium and cesium recoveries are correlated, with the sample showing the highest cesium recovery also showing the highest barium and the sample with the lowest cesium recovery giving the lowest recovery for barium. This relationship between the cesium and barium recoveries is sensible insofar as one would expect the recoveries to be influenced not only by completeness of dissolution but also by the degree to which the analyst was able to wash the solution from the beaker to the dilution vessel. If dissolution and mixing of the solution in the beaker is complete, one would expect the barium-to-cesium ratio, which is key to calculating the age of a 137Cs source, to be unaffected by the extent of transfer, particularly if the fraction of sample transferred is high. The observation that more than 98 % of the Cs in every sample was recovered indicates that the transfer fraction was satisfactory in these tests. The Ba/Cs ratio is larger in the solutions than that calculated from the added amounts, as indicated in the last column of Table 4. This might be due to a trace barium impurity in the starting aluminosilicate or to traces of barium arising from a contaminant in the reagents or the environment where the samples were handled.
Cesium/barium separation
Gas pressurized extraction chromatography (GPEC) operations
In early work on the present task, a miniature GPEC fabricated by INL in 2009 was used for the Cs/Ba separations. Details of the mechanical operation of the GPEC can be found elsewhere [5]. As we proceeded with our study, we began to question the reasons for using the GPEC system in the first place. One of the GPEC features that prompted its adoption in early age-dating applications is its ability to handle small sample quantities, which could reduce the radiation exposure of analysts that do the separations. Other features were a potential for speed in the operations and the possibility of automating the separations process. In our experience using the manual GPEC system, we have not seen its promise realized. For one thing, we have found that the extent of reduction for the sample quantity is not as great as one would expect based on the small loading placed on the column. To fill the sample loop (0.25 mL), we found that one has to have available a much larger volume of sample, at least three times as much, because of the volume of tubing that goes to the loop and the volume needed to ensure complete displacement of liquid that is in the loop and tubing to start. In effect, one ends up throwing away 2/3 of the solution consumed for the separation. The small capacity of the GPEC column is itself a mixed blessing. On the one hand, it allows working with small samples, but on the other hand, when larger samples are available, one cannot use larger amounts to help swamp out contamination effects or benefit from the convenience and accuracy of handling larger portions. Moreover, access to the GPEC systems has not become widespread and only a few of them are in use. The apparatus is quite costly, and, in our hands, has been quite labor-intensive and time-consuming to use. Loop-by-loop elution and collection of the sample fractions takes several minutes per loop with the manual system and if one has to carry out blank separation runs to achieve complete barium recovery, involves considerable analyst effort and potential for higher radiation exposure.
Given all these things, we were open to considering an alternative approach for the separation step when we were invited to participate in an exercise involving age dating 60Co sources [21]. As part of the exercise, we were required to set up a column assembly in a configuration we had not used before. The column resin was packed in an assembly based on a commercially available kit (BenchMark Omnifit Column®, Diba Industries, Danbury, CT). Although it resembles a conventional packed column, the Omnifit® system allowed very convenient handling of solutions and operational control using a peristaltic pump. We were impressed by the ease of use, speed of separations, and excellent separation efficiencies for Co from Ni as well as an ability to achieve quantitative recovery of the separated element fractions. Based on our experience during the 60Co task, we concluded that an analysis process based on an analogous setup might be advantageous for the cesium/barium ceramic case and that it could perform better than the process based on the GPEC system. Experiments to evaluate performance of the Omnifit® column assembly are described in the next section.
Separations using Sr Resin in an Omnifit® column assembly
To evaluate performance of the column with a ceramic-like matrix, an elution profile was created in which we loaded 0.5 mL of a test solution containing nominally 2 ug/g of Cs and Ba, 40 ug/g Na and 50 ug/g Al in 3 N HNO3. Next, 5 mL of 3 N HNO3 was passed through the column to elute Cs and 7 mL of 1 % acetic acid to elute barium. Fractions were collected containing 1-mL increments of the solution that flowed out of the system. Each fraction was weighed to determine the exact amount recovered. The profile obtained with the 50-mm Omnifit® column is shown in Fig. 3.

Profiles of cesium and barium elution with sodium and aluminum present in sample: 50-mm Omnifit column; 0.5 mL sample in 3 N HNO3; washed with 5 mL 3 N HNO3 and then 7 mL 1 % HOAc
Adding aluminum and sodium to the sample did not change the cesium elution characteristics substantially. However, it is clear that the presence of these elements causes barium to be displaced from the resin much earlier in the 3 N HNO3 wash. Baseline separation of cesium and barium still occurs but the collected fractions have to be carefully sized to avoid barium being present in the cesium fraction or cesium in the barium fraction.
For our next test, we processed diluted solutions of the the three spiked ceramics and the unspiked ceramic sample described in Table 2 through the 50-mm Omnifit® column. In each separation, we loaded 0.5 mL of the sample solution (in 3 N HNO3) and rinsed the column with 3 mL of 3 N HNO3 to produce what we designated the Cs Fraction. Then, we changed the collection tube and passed 5 mL of 1 % acetic acid through the column to elute Ba, producing the Ba Fraction. Each fraction was ultimately diluted to 50 mL with 2 % nitric acid for ICP-MS determination Cs and Ba. The column was reconditioned with 5 mL of 3 N HNO3 between samples. Results from analysis of the fractions are summarized in Table 5.
Although the element separations with this Omnifit® column appear to be fairly good insofar as only traces of Cs were found in the Ba fractions and traces of Ba in the Cs fractions, the Ba/Cs ratios calculated from the separated fractions are shifted to values lower than the values from analysis of unseparated portions of the same sample solutions. One possible reason for the low Ba recoveries indicated by this observation is that we did not wash the column with sufficient 1 % acetic acid to remove all the Ba from the column. The elution profile data seemed to make this very unlikely, but we could not discount it. Based on data presented below, we presently believe that the underlying cause is more likely a failure of the Sr resin to completely release barium to the 1 % acetic acid eluent.
Given the small separation window that was found between the cesium and barium peaks with the 50-mm long Omnifit® column, we procured a kit with column dimensions of 3-mm i.d. by 100 mm in length. The 100-mm column was packed in the same manner as the shorter column.
An elution profile obtained with the 100-mm Omnifit® column when we loaded 0.5 mL of sample and then passed 5 mL 3 N HNO3 and 7 mL 1 % acetic acid is shown in Fig. 4.

Profiles of cesium and barium elution with sodium and aluminum present in sample, 100-mm Omnifit column: 0.5 mL sample in 3 N HNO3; wash with 5 mL 3 N HNO3 and then 7 mL 1 % HOAc
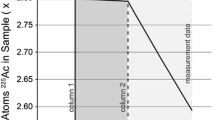
The longer column performed as expected and retained barium throughout the entire wash with 3 N HNO3 (the single point showing an elevated Ba concentration at about 3 g of eluent in the plot is attributed to a spurious contamination and is not considered real). Cesium eluted from the longer column almost as fast as it did from the shorter one; this is not surprising since the cesium washout is essentially determined by the system dead volume and doubling the column bed volume from 0.35 mL to 0.70 mL only increases the dead volume by 0.60 × 0.35 mL = 0.21 mL based on Eichrom’s value of 0.60 for the free-volume fraction of a packed Sr resin bed.
We repeated the tests with the surrogate ceramic samples using the 100-mm Omnifit® column. Results of these tests are given in Table 6.
These tests also show the pattern of low Ba/Cs ratios after separation that we saw in GPEC tests and in the test with the 50-mm Omnifit® column. In all cases, barium recovery appears to be low by about the same fraction, 2 % of the amount loaded on the column. It seemed that the 1 % acetic acid eluent was not extracting all the barium from the column. We therefore began thinking about ways in which we might improve the effectiveness of removing barium from the resin. On looking at the Eichrom specifications for the Sr resin, we noticed that Eichrom recommends eluting Sr and Ba with dilute (0.01 M) nitric acid. So, we decided we would try 0.01 M HNO3 in place of the 1 % acetic acid. The elution profile and surrogate ceramic separation test data with this eluent and the 100-mm Omnifit column are shown in Fig. 5 and Table 7. Almost no difference is seen between performance with the 0.01 M nitric acid elution of barium and that with 1 % acetic acid.

Profiles of cesium and barium elution with sodium and aluminum present in sample, 100-mm Omnifit column: 0.5 mL sample in 3 N HNO3; wash with 5 mL 3 N HNO3 and then 7 mL 0.01 M HNO3
The question remained, “how can one improve the desorption of barium from the Sr resin?” Two possibilities came to mind. One would be to decrease the acidity of the solution since the distribution coefficient (K d) for Ba on the Sr resin is known to decrease exponentially with nitric acid concentration. Just using a pH higher than that of 0.01 M HNO3 seemed unlikely to be successful. Even going to pure water as eluent did not seem likely to produce the change we desired since this had been reported by Sommers et al. [1], to be less effective than 1 % acetic acid in their GPEC operations. The other would be to add a complexant to the barium eluent to promote barium back extraction. On searching the literature for Sr resin applications, we found no reports of complexants having been used for stripping Sr resin columns of Ba or Sr. However, we did find a few references [22–24] that described using a dilute solution of ethylenediaminetetracetic acid (EDTA) or cyclohexylenediaminetetraacetic (CDTA) to strip barium from cation exchange columns in separating it from radium. We selected a dilute solution of EDTA to test the concept of using a complexant in the barium eluent to improve recovery. Because the formation constant for the Ba-EDTA complex is relatively small (log Kf ≈ 7.9) [25], complexation is most effective at high pH where H + does not compete effectively with Ba for the complexant. We chose an eluent consisting of 0.05 M disodium EDTA (100 times the anticipated concentration of Ba in the eluent solution) in water, which had a pH of 6.
Using a freshly prepared 100-mm column of Sr resin, we replicated our experiments to separate the Cs and Ba in the four surrogate ceramic sample solutions two times using the EDTA eluent for Ba. Results are shown in Table 8. (We found that our 0.05 M EDTA solution contained a trace impurity of barium, about 0.5 ng/mL, and corrected for it in the barium data shown.) One time, we loaded 0.5 mL of the sample and washed the column with 5 mL of 3 N HNO3 to produce the Cs fraction. Then, we washed the column with 7 mL of 0.05 M EDTA to generate the Ba fraction. The next time, we again loaded 0.5 mL of sample and washed with 7 mL of 0.05 M EDTA to elute barium, but we only used 3 mL of 3 N HNO3 for the wash in the Cs fraction because it appeared some barium might have broken through into the Cs fraction in some of the first-series samples where a 5-mL wash was used. Between samples, we reconditioned the column with 5 mL of 3 N HNO3.
The data from these cursory tests of the EDTA-based eluent for barium give considerable reason to be encouraged that this approach could resolve the shortcomings we observed with 1 % acetic acid as eluent. The average Ba/Cs ratio after separation for the six spiked-surrogate ceramic samples relative to the unseparated samples is 0.994 with a standard deviation of 0.008. This is much improved from the corresponding figure of merit with 1 % acetic acid, which averaged only 0.98 or less. And, this improvement does not take into account that the EDTA separation conditions are not optimized. One refinement suggested by the data in Table 8 is a fine tuning of the volume used for the 3 N HNO3 wash in the Cs fraction. The data hint that using 5 mL for this wash causes a small amount of Ba to break through into the Cs fraction, while cutting back to 3 mL seems not to completely wash Cs out of the system, since a trace of Cs shows itself in the Ba fractions. Some volume between these values is likely to be better. We suspect that some of the variability in the barium recoveries might arise from an insufficient wash with the 0.05 M EDTA eluate. Increasing the EDTA wash would be interesting to test.
Unfortunately, re-inventing the Cs/Ba separation methodology was beyond the scope of the current task, which was undertaken to adapt existing methods for age-dating 137Cs to accommodate the ceramic matrix. The problems we have found with the separation method are not due to constituents in the dissolved ceramic and almost certainly affect all other matrices that have been previously investigated, including CsCl and Cs-loaded glass forms (not to mention the Sr/Zr separations needed in age-dating sources containing 90Sr). Our studies do indicate that, for example, the sodium and aluminum components in our ceramic surrogates affect the separation by slightly lowering the retention of barium on the Sr resin, but this effect is minor and easily accommodated by adjusting column size and/or the volumes of wash solutions used in eluting the Cs and Ba fractions. The Cs/Ba separation is projected to work just as well for the ceramic matrix as it does in these other cases. Given this situation, we did not go further in optimizing the separation under the present task.
This is not to say that we consider the present situation acceptable. We strongly recommend studies to simplify and refine the methodology for separations on Sr resin to make them more widely accessible (e.g., by not requiring the GPEC apparatus) and to eliminate the losses that we have documented when 1 % acetic acid is used as the stripping eluent for the alkaline earths (Ba or Sr). The work we have described can serve as a foundation on which such studies can build. Areas that we believe should be looked at are: (1) Use of the Omnifit ® column configuration as an alternative to the GPEC for separations. We have found the commercially available Omnifit® assemblies to be relatively inexpensive and uncommonly convenient to use. They are well-suited to samples containing microgram amounts of 137Cs, only a few times more than that required for the GPEC, but can be easily sized for larger samples if desired. We looked at 3-mm-i.d. micro-bore columns that were 50- and 100-mm long; a 150-mm length that would allow considerably better separation of Cs and Ba with little penalty in time or waste volumes is available and should be evaluated. (2) Refinement and optimization of alkaline earth elution from Sr resin using a complexant-based eluent. In our scoping tests, we tried only one candidate complexant to promote complete removal of barium from the Sr resin. The EDTA complexant we used has a relatively small Kf with barium (log Kf is 7.9 as cited earlier). In other applications, workers reported adjusting the pH of the EDTA solution, using ammonia for example, to enhance the extent of complexation. Increasing the pH of the EDTA eluent could be beneficial and should be investigated. Alternatively, using a stronger complexant like CDTA (log Kf with Ba = 8.6) might also be advantageous. Such improvements promise not only to enhance recovery for barium from the Sr resin, but also to resolve difficulties reported for separations involving strontium. Extending the concept of complexant-based desorption to the Sr/Zr separation required for age dating 90Sr sources might very well reduce the variability in recoveries that made isotope dilution necessary for accurate age dating of that radioisotope [5]. Although using the isotope dilution approach is very effective, it entails considerable penalties in required facilities, effort, time, and cost that are very unattractive in a forensic application. Pursuit of a simpler, more effective process for achieving reproducible and reliable separation and recovery of Sr as well as Ba using the Sr resin, whether using a GPEC or conventional column configuration, seems to us to be a very wise investment.
Projection of 137Cs age-dating method performance
In the following method-performance evaluation, the principles of uncertainty propagation are applied in assigning a value for the uncertainty associated with the “time since purification.” To begin the process, one needs to lay out the individual procedural steps in the determination and describe them using mathematical formulas that explicitly show the dependencies between outcomes and inputs and allow estimation of the effects of all component uncertainties in the input variables so their effect on the outcome can be evaluated [6].
In the present case, we are at a bit of a disadvantage in this endeavor because the work done in our study did not apply an integrated, defined procedure for the age determination, but rather, investigated several procedural concepts pertinent to various steps in the age-dating process. To permit our projecting a method performance, we take the approach of using a postulated method composed of operations that we see as practical and efficient with predictable uncertainty.
Our model method for age dating a 137Cs-ceramic source begins with dissolving the source material. The dissolved sample is transferred to an appropriate vessel and diluted with 3 N nitric acid to prepare a well-mixed stock solution containing the 137Cs parent and 137Ba daughter from the source sample. A portion of this stock solution is diluted by mass to provide a working solution that contains 137Cs at a concentration appropriate for use in subsequent separation and measurement steps. To determine what this concentration should be, we need to consider available measurement and separation strategies that might be applied and anticipate quantities of the analytes of interest that will produce a sample suitable for their application. Our consideration of the measurement and separation aspects of the method warrants discussion at this point because it leads to some insights that help choose between options and facilitate our evaluation of performance.
It has been noted that there are two options for measuring the 137Cs isotope in solution: either radiometric (gamma spectrometry) or mass spectrometric (ICP-MS). In general, the mass spectrometric approach has been favored by researchers who have done 137Cs age dating in the past—likely because their method development (like ours) was done using non-radioactive 133Cs, which requires a mass spectrometric measurement, as a surrogate for 137Cs. Measuring the radioactive 137Cs and the non-radioactive barium with the same instrument also has advantages in convenience and cost. When we laid out a detailed concept for the operations to determine 137Cs by ICP-MS, however, it came to light that a serious gap exists in the processes needed for what we would consider a satisfactory calibration of the system. To obtain an accurate calibration, one wants ideally to calibrate the ICP-MS response using the specific isotope that will be measured. This is necessary because mass spectrometric measurements are subject to a mass discrimination effect that causes different isotopes of the same element to exhibit different detector responses for identical concentrations. When laboratories have used ICP-MS for measuring 137Cs, they accommodated this effect by assigning the same sensitivity for 137Cs as was observed for 137Ba or for 133Cs. Such actions are common in ICP-QMS operations in cases where specific isotope standards or isotopic reference materials are not available and where the uncertainty introduced by the assignment (typically presumed to be < 10 %) is tolerable for the application. It was our judgment that these approaches are not acceptable for the age-dating method. One option that came to mind to circumvent this problem was to use the 137Cs in the NIST SRM 4233E (Cesium-137 Radioactivity Standard) for calibrating the ICP-MS. A problem with this option is that the cesium in this standard would have to be separated from the barium component. This would require not only the effort to carry out the separation, but also radiometric measurements to evaluate recovery of the 137Cs in the separation step. It is clear that a better option would be to measure the 137Cs amount in the age-dating sample using gamma spectrometry, which could be directly calibrated against the NIST SRM. This not only provides an accurate calibration and establishes traceability, but also allows uncertainty components in the measurement to be assigned in a rigorous framework since the standard-sample comparison using a gamma counter is well understood. In our model age-dating method, then, we specify that the 137Cs component in the sample will be determined using gamma spectrometry. We note that the 137Cs measurement in this case can be made either before or after separating the cesium from barium since barium in the sample does not interfere with the gamma measurement; if the measurement is made after separation, additional measurements should be made to evaluate recovery of the cesium component during the separation process.
By necessity, the barium component of the age-dating sample is measured by ICP-MS in our model scheme. Calibration of the ICP-MS for 137Ba is achieved using solutions prepared from a NIST traceable standard containing natural barium and the assumption that barium in the standard has an isotopic composition that falls in the bounds of terrestrial variations listed by IUPAC [20] (See Table 9).
This approach provides direct calibration of the 137Ba signal with standards having documented uncertainty sources.
We have described two possible approaches to separating barium from a measured amount of the working solution prepared from the 137Cs source—separation with the GPEC system and separation with the Omnifit® column assembly. To simplify comparison of these separation techniques, we postulate using an amount of solution that contains 1 µg of 137Cs, equivalent to 3.212 MBq of 137Cs activity, in both cases. With the GPEC system, this quantity of 137Cs would be contained in a volume of sample solution that corresponds to one sample loop (typically 0.25 mL). We recognize that the activity of this solution might be higher than desirable for operations in some facilities but we also believe that lower concentrations can be used without substantively affecting method-performance conclusions based on this working value. In our postulated method using the Omnifit® apparatus, we envision loading 0.5 g of sample solution to the column, with the mass of added solution measured when the loading is done (we did this in our tests by weighing the vial containing the load solution before and after the solution was pumped to the column); again, a smaller volume or somewhat lower concentration will have minimal effect on the performance outcome as long as the solution mass is large enough to allow precise weighing and the barium amount in the separated fraction is sufficient to allow measurement with uncertainties consistent with postulated conditions.
In visualizing operations for the separation processes, we recognized two procedures that might be followed to evaluate the ratio of 137Ba/137Cs in the source material. In one embodiment of the process, the 137Cs concentration of the sample solution would be measured prior to separation while the 137Ba concentration would be determined from the mass of solution loaded to the column and the amount of 137Ba measured in the separated barium fraction, with correction for recovery of barium during the separation. In the second embodiment, both the 137Cs and the 137Ba amounts in the separated fractions would be determined in the respective separated fractions using measurements carried out on solutions in which the fractions would be contained. The 137Cs would be measured by gamma counting, and the 137Ba by ICP-MS. Again, recovery corrections might be necessary. In this second case, it is not necessary to know the exact amount of solution used in the separation since it cancels when the isotope ratio is calculated. The first approach has some advantage when used with the Omnifit® system since it allows convenient measurement of the 137Cs concentration and the mass of separated solution is easily and accurately measureable. The second approach is advantageous with the GPEC system since the amount of solution in the sample loop has to be deduced from a calibration of the sample loop and the density of the sample solution has to be known or measured for calculations. These requirements entail added effort and introduce added uncertainty, which can be avoided by measuring the isotopes in their respective fractions. Both processes involve carrying out dilutions, additions of internal standard (for barium), and other manipulations of solution samples, but these can be done by weight with very small uncertainty (<0.05 %, relative).
There is one additional point to be made in laying out the age-dating method considerations. It concerns the interchangeability among units for expressing factors in the ratio of 137Ba/137Cs amounts, which forms the basis for calculating the age of a source. In Eq. 2, an expression is presented that involves the ratio of atom amounts, \( N_{{^{137} {\text{Ba}}}} \left( t \right)/N_{{^{137} {\text{Cs}}}} \left( t \right) \). We note that, because the nuclidic masses of the two isotopes are virtually identical (to better than 0.01 % of their values), this atom amount ratio is numerically equivalent from a practical standpoint to the corresponding ratio where the elements are expressed in mass quantities, \( M_{{^{137} {\text{Ba}}}} \left( t \right)/M_{{^{137} {\text{Cs}}}} \left( t \right) \). Moreover, if the two isotopes are present together in solution, the ratio of concentrations is also identical to the ratio of atom amounts because the solution-mass or solution-volume component of the concentration expression cancels when the ratio is taken. Because of these identities, it does not matter whether the 137Cs and 137Ba values are expressed in terms of atom amounts, mass amounts, concentrations per unit mass of solution, or concentration per unit volume of solution; the ratio will have the same value as long as the two isotopes are presented on the same basis. For this reason, we do not take time to detail conversions implicit in switching from one set of units to another in our mathematical treatments of the age-dating process.
Uncertainty propagation calculations are based on the expression in Eq. 4, which is derived from a Taylor Series expansion of the formula that describes the relationships among variables used in determining a system property [6].
In this expression, σ 2(y) represents the variance of the property y and is given as the summation of the products of sensitivity coefficients, c 2(i), and variances, σ 2(i) for individual variables, i. The sensitivity coefficient weights the variance from each variable according to its effect on the variance in y and is obtained by partial differentiation with respect to i of the expression that relates the variable to the property.
When the summation in Eq. 4 is composed using standard uncertainties expressed as a standard deviation for the pertinent variables, \( \sigma \left( y \right) \) is called a combined uncertainty. Method performance is commonly rated in terms of the expanded uncertainty, calculated as the product of the combined uncertainty for the measured property and a coverage factor, k, which corresponds to a specified confidence interval around the reported value of the property. Ellison et al. [6] recommend using a coverage factor of 2 in calculating the expanded uncertainty.
For our calculations, we started with an expression (Eq. 6) for the age parameter, t, in a slightly different format than that presented earlier in Eq. 2. This formula is written in terms of the half-life, t 1/2, rather than the decay constant, λ. The two expressions are identical since, by definition, λ = ln(2)/t 1/2.
Here, R represents the 137Ba to 137Cs amount ratio calculated from atom amounts, mass amounts, or concentrations of the two isotopes in the source sample.
Using the strategy implicit in Eq. 4 to evaluate sensitivity coefficients for the pertinent variables, uncertainty in t is related to uncertainty in the half-life and in the measured value of R as in Eq. 7.
The partial derivatives in this expression have the values shown in Eqs. 8 and 9.
These expressions indicate that the uncertainty in the assigned age value is not a constant, but will depend on the value of R and, consequently, vary with the magnitude of the determined age.
The half-life of 137Cs has the value 30.070 ± 0.030 year [26], where the uncertainty shown is the standard uncertainty. This information is sufficient for one to evaluate the first term in Eq. 7 for any value of R.
The quantity R is a function of other variables evaluated in the course of processing the sample, and the second term must be expanded before it can be used in treating the uncertainty in t. In our model system, we have postulated that a sample of the 137Cs ceramic source is dissolved, the resulting solution is diluted to an appropriate concentration, the 137Cs concentration is measured by a gamma-counting comparison with standards prepared from NIST SRM 4233E, barium in the solution is separated from cesium using Sr-resin in a GPEC or Omnifit® apparatus, and the barium concentration is measured using ICP-Quadrupole Mass Spectrometry (ICP-QMS) with non-linearity and mass-discrimination effects corrected using the Argonne model algorithm [5]. The formula we composed to describe our determination of R with this methodology is given in Eq. 10.
In this formula, F Diss represents a factor that accounts for any shift in the ratio value that might occur as a result of incongruent dissolution of the cesium and barium constituents in the ceramic sample or any segregation of the elements due to incomplete sample transfer or incomplete mixing of the sample stock solution. Based on the discussion of the dissolution procedure in “Results and discussion”section, we assign the value unity to F Diss with a relative standard uncertainty of 0.l % to account for effects that might be present and were not recognized in our testing.
The factor F Dil represents a factor that accounts for dilutions that are carried out during the dissolution operations, including the initial serial dilution of the sample stock solution. Terms contributing to this factor will depend on the exact sequence of dilutions applied in processing the samples, but are expected in the end to cancel when the cesium- and barium-related contributions are normalized to a particular condition used in evaluating the concentrations; thus, we also assign the value unity to F Dil. Because we apply a strategic design to the dilutions and ensure that the amounts of solution are always sufficient to allow precise and accurate measurements of their mass, we assign a relative standard uncertainty of 0.05 %.
The factor F Sep corrects for any shift in the ratio value that occurs because of differences in the recovery of barium and cesium in the separation process. As discussed earlier, evidence exists that this factor has a value less than unity for the GPEC system. With the Omnifit® apparatus, the value might be close to unity, although additional development and characterization is needed to establish what the optimized value might be. For the sake of the present analysis, we elected to use the data from our investigations of the two separation techniques. We assign for F Sep using the GPEC a value of 0.983 with a standard uncertainty of 0.023 (2.3 % relative) [4]. With the Omnifit® system and an EDTA eluent, the corresponding value is 0.994 with a relative standard uncertainty of 0.8 % (Table 8). Because the value of F Sep is expected to vary randomly from sample to sample, the magnitude of the uncertainty term corresponding to F Sep is reduced by the factor N Sep if replicate sample portions are put through the separation and measurement process.
The variable \( C_{{^{137} {\text{Ba}}}} \) represents the concentration of 137Ba measured in the solution analyzed by ICP-MS and normalized to the same condition as the solution to which the 137Cs measurement is calculated. The ICP-MS measurement involves comparing signals from the sample to those from standards prepared by diluting a certified spectroscopic standard solution. A simplified expression describing the measurement is given in Eq. 11.
Here, S Sple and B Sple are the instrument signal and the subtracted background, respectively, for the sample. The quantity Slope is the slope of the calibration curve, which is derived from measurements with the working standards, and is expressed as in Eq. 12.
S Std is the signal recorded for the standard and C BaStd is the concentration assigned to the working standard solution. C BaStd is calculated as described earlier from the certified concentration of barium in the reference solution, C 0BaStd , the mass fraction abundance of 137Ba in natural barium, \( A_{{{\text{Nat}},^{137} {\text{Ba}}}} \), and a dilution factor, DF BaStd, derived from mass measurements recorded when the working standard is prepared.
The last quantity needing consideration in Eq. 10 is the variable \( C_{{^{137} {\text{Cs}}}} \), which represents the measured concentration of 137Cs in the solution that corresponds to the solution analyzed for 137Ba with the ICP-MS. This concentration is determined by a gamma-counting comparison of an appropriate dilution of the sample solution with standards prepared from the NIST SRM 4233E.
In this expression, CR Sple and CR γStd are the count rates recorded during gamma counting of the sample and the NIST SRM 4233E standard, respectively. C 0NIST,γ is the concentration of 137Cs in the NIST SRM (available from the NIST certificate), and DF γ is a factor that accounts for dilutions made in preparing the sample and standard solutions for counting.
The relationships in Eqs. 6–14 identify fourteen input quantities that contribute uncertainty to the age measured for a source of interest. For each of these, an expression for the sensitivity coefficient was derived and a standard uncertainty was assigned. The sensitivity-coefficient expressions are listed in Table 10; standard uncertainties are presented in Table 11 along with a statement of the method we used for evaluating each uncertainty.
Based on the information available from the foregoing discussion, an expression was derived that provides the desired combined uncertainty in t as a function of the uncertainties in the input quantities.
In this expression, the conventional symbols are used to represent the pertinent standard uncertainties [u(i) is the standard uncertainty in the quantity i; u r(i) is the relative uncertainty in i, evaluated as u(i)/i and expressed as a fraction; u c(t) is the combined uncertainty in the measured age].
One feature of this expression that warrants comment is the appearance of the quantity \( C_{{^{137} {\text{Cs}}}} \) in the third term. This value of the 137Cs concentration corresponds to the 137Cs that would be present in the solution analyzed for 137Ba if it had not been separated out. The exact value of this quantity depends on the details of the operations and dilutions carried out in processing the source sample. For our treatment, we chose to set the value at 20 ng 137Cs per g solution, which we consider a mid-range target value in our operations. Since this last term only contributes consequentially when the age of a sample (or, equivalently, the value of R) is very small, the effect of choosing a smaller or larger concentration has little influence on conclusions we draw from our calculations.
Using the expression in Eq. 15, we constructed uncertainty budgets corresponding to several conditions of measurement, including variations in the separation applied (GPEC versus Omnifit®), the value of R (which establishes the measured age of the source), and the number of replicate separations (N sep) or ICP-MS measurements (N ms) that were done. Each budget lists the input quantities and the values of the terms that include the input’s standard uncertainty, which represent the contributions of each input to the combined uncertainty in t. Indexing each variance term as a percentage of the total variance in t provides a measure of the relative importance of each input uncertainty for determining the total. Uncertainty budgets for operations using the GPEC and the Omnifit® separations are summarized in Tables 12 and 13, respectively. In each case, N sep = N ms = 3; values are shown for R = 0, 0.5, 1, 2, and 3 to illustrate the magnitude of the uncertainty for different age results.
The condition where R = 0 represents a special case in the treatment insofar as 100 % of the combined uncertainty in t arises from the signal noise in the ICP-MS measurement of 137Ba. In this case, we project an age uncertainty of about 0.015 year (expanded).
When R is consequentially larger than 0, regardless of whether the GPEC or Omnifit® separation is used, the largest contributor to uncertainty in the measured age is the variability in F Sep, which is related to the reproducibility of barium recovery during the separation. With the GPEC system, this factor accounts for about 85 % of the variance in t. Its fractional contribution is considerably smaller (40 %) with the Omnifit® arrangement using an EDTA eluent. The expanded uncertainty in t is only about half as large with the Omnifit® arrangement as with the GPEC.
The next largest contributor to uncertainty in the measured age is uncertainty in the certified concentration of 137Cs in the gamma calibration standard. Reducing that uncertainty, and for that matter, uncertainties in most other input variables, is not simple and not likely to produce more than incremental improvements in the overall uncertainty in the measured age. There does appear to be an opportunity to make small gains in the method performance by improving the separation procedure. If variability in the separation recovery could be reduced from the 0.8 % rsd estimated from our Omnifit® data to negligible levels, the expanded uncertainty in the measured age would be reduced to about 75 % of the values listed in Table 13. The uncertainty reduction produced by replacing the GPEC with the Omnifit® apparatus using a complexant-based eluent is evident in this result. With the performance we project with the Omnifit® approach, the age of even the oldest 137Cs source (about 60 years) can be determined with an expanded uncertainty of less than one year.
Conclusions
The methodology used for age dating radioactive sources containing 137Cs as chloride salt or glass was adapted for sources where the 137Cs is incorporated into a ceramic matrix. Using a surrogate cesium aluminosilicate, we evaluated potential effects of the matrix on the individual processes needed for applying the age-dating method, including dissolution of the matrix, separation of the cesium and barium constituents, and measurement of the 137Cs and 137Ba isotopes.
In general, effects of the ceramic matrix were minor. The aluminosilicate material was readily dissolved with a procedure that incorporated hydrofluoric acid and subsequent removal of excess fluoride. The resulting solution provided high recovery of the cesium and barium components and maintained the Ba/Cs ratio during handling and transfers. The matrix constituents, sodium and aluminum, did not affect the Cs/Ba separation measurably with the GPEC apparatus but barium retention was clearly reduced during elution of the cesium with 3 N HNO3 when the separation was done using more conventional chromatography and an Omnifit®-column setup. This effect could be readily accommodated by using a longer column and selecting an appropriate volume for eluting the cesium fraction. No effects were observed in the ICP-MS measurements of Cs or Ba from the sodium and aluminum present in the sample solutions. Overall, age dating of 137Cs incorporated into an aluminosilicate ceramic matrix is anticipated to be just as reliable as age dating this isotope in any matrix that was previously studied.
For all matrices, the age-dating methodology for 137Cs can be substantially improved by changes to the procedures for separating the cesium and barium isotopes and for measuring the 137Cs constituent. The manual GPEC has not realized expectations for advantageous performance and GPEC systems have not become widely available as once anticipated. Consequently, we recommend an alternative apparatus based on the readily available and inexpensive Omnifit® column kit. This approach allowed much more convenient operation, better utilization of sample, and a potential for more control over separation conditions than the GPEC. With either system, recovery of barium in a fraction eluted with 1 % acetic acid was consistently low by about 2 % and varied substantially from one run to another (average recovery was 98.3 % with a standard uncertainty of 2.3 %). A short study of alternative eluents for the barium fraction found that adding a complexant such as EDTA in place of acetic acid improved the recovery (99.4 ± 0.8 %). This improvement constitutes a major reduction in the overall uncertainty of the age-dating method since variability in the barium recovery is the largest single contributor to the combined uncertainty in the measured age, as illustrated in the uncertainty analysis we present.
Consideration of the procedures available for measurement of the 137Cs isotope for the age-dating process revealed a serious gap in the chain of comparisons that underlie calibration of the ICP-MS instrument for this nuclide. Assumptions commonly made in earlier work regarding identity of responses between 137Cs and 133Cs or 137Ba cannot be justified within reasonable uncertainty. This impediment is removed if the 137Cs determination is made radiometrically using gamma counting where suitably traceable standards are available and measurements can be made with predictable uncertainty.
With the recommended methodology, uncertainty in the age of a 60-year-old source is 0.68 y.
References
Sommers J, Cummings D, Giglio J, Carney K (2009) Age determination of irradiated materials utilizing inductively coupled plasma mass spectrometric (ICP-MS) detection. J Radioanal Nucl Chem 282:591–595
Goldberg M, Chamberlain D, Kalensky M, Lewis M, Mertz C, Tsai Y (2009) Age-Dating and Analysis of ‘Ames’ 137Cs Sealed Source Material, Final Report. Argonne National Laboratory Technical Report ANL-NTNFC-09-0022
National Research Council Committee on Radiation Source Use and Replacement (2008) Radiation sources in the United States and their uses and origins: abbreviated version. National Research Council of the National Academies, National Academies Press, Washington, D.C
Steeb JL, Mertz CJ, Sandi G, Bass DA, Graczyk DG, Goldberg MM (2012) Micro-separations of cesium and barium in glass. J Radioanal Nucl Chem 292:757–762
Steeb JL, Graczyk DG, Tsai Y, Mertz CJ, Essling AM, Sullivan VS, Carney KP, Finck MR, Giglio JJ, Chamberlain DB (2013) Application of mass spectrometric isotope dilution methodology for 90Sr age-dating with measurements by thermal ionization and inductively coupled plasma mass spectrometry. J Anal At Spectrom 28:1493–1507
Ellison SLR, Rosslein M, Williams A (eds) (2000) Quantifying uncertainty in analytical measurements, Eurachem/CITAC Guide CG 4, 2nd edition QUAM:2000.1
Dejsupa C, Heo NH, Seff K (1989) Crystal structure of cesium zeolite A prepared by complete aqueous exchange. Zeolites 9:146–151
Schultz FJ, Tompkins JA, Haff KW, Case FN (1981) Preparation and characterization of cesium-137 aluminosilicate pellets for radioactive source applications. Oak Ridge National Laboratory technical report ORNL-5775
Vereshchagina TA, Vasilieva NG, Vereshchagin SN, Paretskov EN, Zykova ID, Krucheck DM, Manakova LF, Tretyakov AA, Anshits AG (2006) Porous materials based on cenospheres of coal fly ash for fixation of 137Cs and 90Sr in mineral-like aluminosilicates. Mater Res Soc Symp Proc Vol 932
Kaminski MD, Mertz CJ, Ferrandon M, Dietz NL, Sandi G (2009) Physical properties of an aluminosilicate waste form for cesium and strontium. J Nucl Mater 392:510–518
Enomoto S, Maeda S, Senoo M (1989) A simplified method for preparation of 137Cs pollucite γ-ray source. Int J Appl Radiat Isot 32:595–599
Milearski M (1989) Preparation of 137Cs pollucite source core. Isot Environ Health Stud 25:404–408
Scheetz BE, Aggarwal DK, Breval E, Roy R (1994) Sodium Zirconium Phosphate (NZP) as a host structure for nuclear waste immobilization: a review. Waste Manag 14:489–505
Chourasia RB, Ambastha RD, Shrivastava OP, Wattal PK (2010) Crystallographic evaluation of sodium zirconium phosphate as a host structure for immobilization of cesium. J Mater Sci 45:533–545
Richerson DW (2006) Modern ceramic engineering: properties, processing, and use in design. CRC Press, Boca Raton, FL
Schlesinger HI, Brown HC, Mayfield DL, Gilbreath JR (1953) Procedures for the preparation of methyl borate. J Am Chem Soc 75:213–215
Salit ML, Turk GC (1998) A Drift Correction Procedure. Anal Chem 70:3184–3190
Salit ML, Vocke RD, Kelly WR (2000) An ICP-OES method with 0.2 % expanded uncertainty for the characterization of LiAlO2. Anal Chem 72:3504–3511
Salit ML, Turk GC, Lindstrom AP, Butler TA, Beck CM, Norman B (2001) Single element solution comparisons with a high-performance inductively coupled plasma optical emission spectrometric method. Anal Chem 73:4821–4829
Berglund M, Wieser ME (2011) Isotopic compositions of the elements, 2009. Pure Appl Chem 83:397–410
Charbonneau L, Benoit JM, Jovanovic S, St-Amant N, Kiser S, Cooke MW, Mercier JF, Nielsen K, Kelly D, Samuleev P, Galea R, Moore K, Saul PRB, Chamberlain DB, Steeb JL, Graczyk DG, Tsai Y, Sullivan VS, Dimayuga IC, Shi Y, Rao R, Lariviere DA (2014) Nuclear forensic method for determining the age of radioactive cobalt sources. Anal Methods 6:983–992
Nelson F (1964) Ion exchange procedures: V. Separation of barium and radium. J. Chromatogr 16:403–406
Ditchburn RG, Whitehead NE (1995) Separation of Ra and Th from rock matrices for alpha-spectrometry. J Radioanal Nucl Chem 189:115–125
Gleason G (1980) In: Lyons W (ed) Radioelement analysis progress and problems. Ann Arbor Science, Ann Arbor
Reilly CN, Schmid RW, Sadek FS (1959) Chelon approach to analysis: I. Survey of theory and application. J Chem Ed 36:555–564
Evaluated Nuclear Structure Data File (ENDSF), September 2005
Acknowledgments
Argonne National Laboratory’s work was funded by the U.S. Department of Homeland Security. The submitted manuscript includes information created by UChicago Argonne, LLC, operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and display publicly, by or on behalf of the Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Steeb, J.L., Graczyk, D.G., Tsai, Y. et al. Age-dating methodology for 137Cs ceramic sources. J Radioanal Nucl Chem 309, 999–1019 (2016). https://doi.org/10.1007/s10967-016-4712-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4712-x